> ## Documentation Index
> Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
> Use this file to discover all available pages before exploring further.
# Therapeutic Strategy and Hit Matching
> Identify Hits That Match a Specific Binding Modality or Mechanism
## **The Problem You Are Trying to Solve**
*“I have a biological target, and I want to identify a set of hits that meet a specific mechanistic or chemical criterion, like specific modes of action: inhibition, activation, covalent binding, or other specialized mechanisms.”*
 At hit identification, not all hits are equal. Different discovery goals require fundamentally different binding behaviors, and a generic virtual screen often fails to distinguish them. Common challenges include:
* Docking scores that don’t reflect functional outcome (inhibition vs activation)
* Difficulty identifying covalent or mechanism-based binders
* Binding poses that look plausible but don’t explain functional modulation
* Mixing multiple binding modalities in a single hit list
This workflow is designed to explicitly tailor hit identification to your intended mechanism of action, producing hits that are aligned with *how* you want the target to be modulated.
## **Solution**
This workflow uses Revilico’s binding chemistry engines to customize hit discovery based on binding modality, combining structural filtering, interaction analysis, and (when needed) dynamic or energetic validation. The shared core workflow is: Target Preparation → Mechanism-Aware Docking → Interaction & Motif Analysis → Hit Prioritization.
Where the workflow diverges is in:
* How docking is configured
* What interactions are emphasized
* Which downstream validation steps are applied
Integration with MD, FEP, QSAR, and generative chemistry is possible once modality-specific hits are identified, and the campaign has reached an iterative optimization stage.
## **What Data Do I Need to Provide?**
Required
* Target protein structure (experimental or predicted)
* Compound library (or libraries) to screen
Recommended
* Known ligands or reference compounds for the desired modality
* Information about active sites, allosteric sites, or reactive residues
Optional
* Cofactors, ions, or partner proteins
* Experimental benchmarks for validation
## **Workflow**
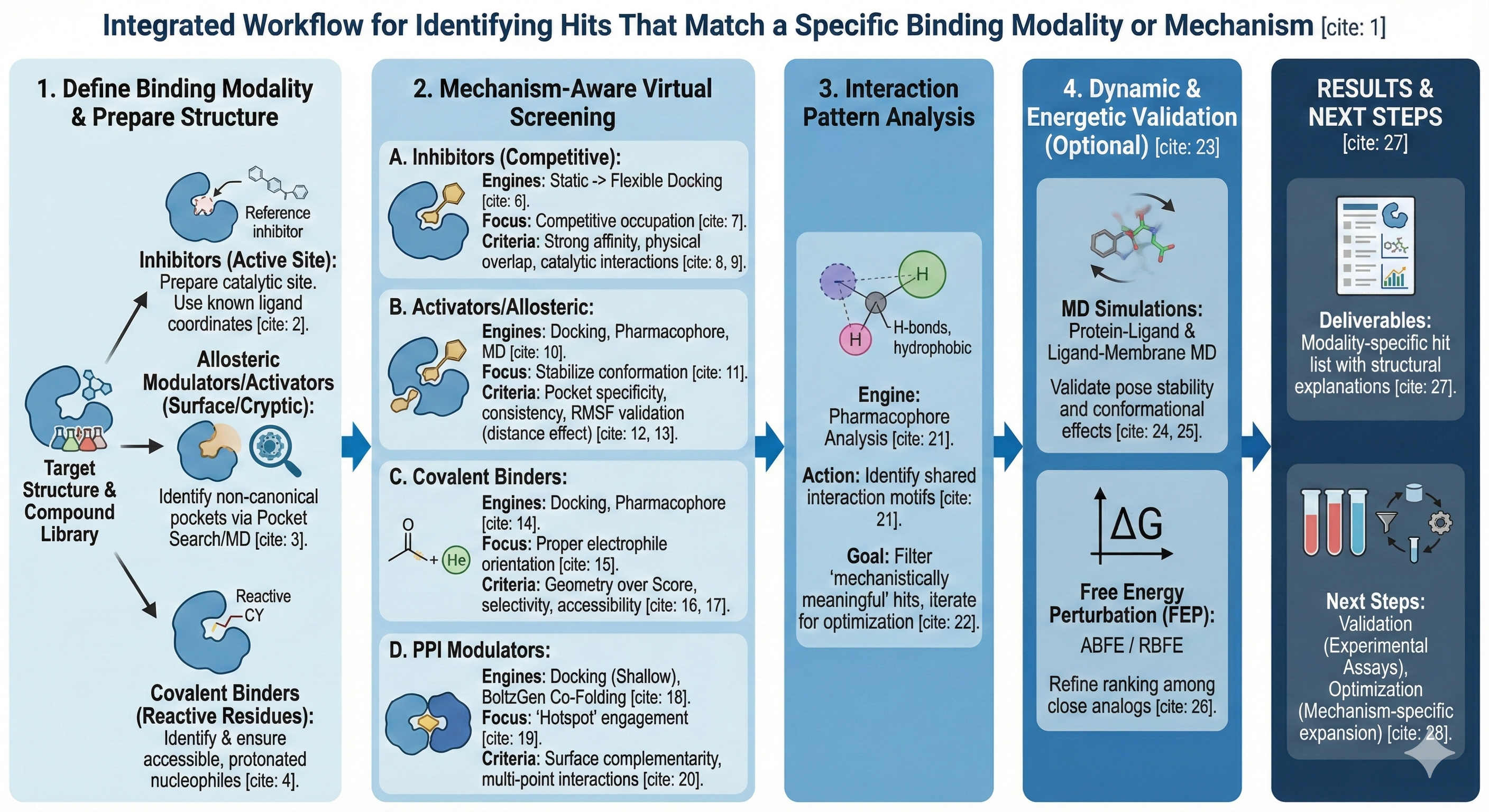
1. **Define the Desired Binding Modality**
Before screening, explicitly define *what kind of hit you want*.\
Common criteria include:
* Inhibitors (competitive, non-competitive)
* Activators / positive allosteric modulators
* Covalent binders
* Allosteric binders
* Stabilizers / disruptors of protein–protein interactions
* Membrane-active or permeability-driven binders (when relevant)
This decision informs every downstream configuration choice to ensure that binding and engagement are translating to biologically relevant functions.
2. **Prepare Target Structures Based on Modality**
Different modalities require different structural contexts.
* Active-site binders (inhibitors):
* Prepare catalytic or orthosteric site
* Use known substrate or inhibitor coordinates when available as a reference
* Allosteric modulators / activators:
* Identify non-canonical pockets via literature, pocket detection, or prior MD
* Multiple conformations may be needed
* Covalent binders:
* Identify reactive residues (e.g., Cys, Ser, Lys)
* Ensure correct protonation and residue accessibility
Primary engines used
* **Docking** (for pose hypothesis)
* Optional **Protein Water MD** (to reveal transient or cryptic pockets) with potential follow up by the Pocket Search Engine to identify pockets of interest.
This step will give you one or more docking-ready target representations aligned with the mechanism.
3. **Mechanism-Aware Virtual Screening**
#### **A. Inhibitor Discovery**
Primary engines
* **Static Docking** → **Flexible Docking** (for refinement)
Key focus
* Competitive occupation of known binding pocket
* Strong, well-oriented interactions with catalytic residues
* Consistent low-energy poses
What you prioritize
* Affinity trends, especially hits that are reading out lower/negative kcal/mol energies
* Pose stability, looking at physically feasible binding and lack of steric clashes
* Overlap with known inhibitor binding modes, attaching to key amino acids of interest in the pocket
#### **B. Activators / Allosteric Modulators**
Primary engines
* **Docking** (alternative pockets)
* **Pharmacophore Analysis**
* **Molecular Dynamics Simulation Protein Ligand and Protein Water for Calibrations**
Key focus
* Binding outside the active site
* Interactions that stabilize specific protein conformations
* Poses that plausibly alter protein dynamics, and can be measured through molecular dynamics and Root Mean Squared Fluctuation (RMSF) values
What you prioritize
* Pocket specificity, ensuring that you are engaging with the right allosteric pocket
* Interaction networks rather than raw affinity scores, and physically feasible conformations
* Consistency across protein conformations
Validating the results
* In order to validate that your designed compound is an allosteric modulator, you can put the pose into molecular dynamics simulation over longer time span and look for RMSF value increases across longer time scales, in the active site of the given protein.
* Your designed compound should reveal a larger fluctuation pattern in the protein after ligand engagement compared to just protein in water MD
#### **C. Covalent Binder Identification**
Primary engines
* **Docking** (pose feasibility)
* **Pharmacophore Analysis** (reactive geometry validation)
Key focus
* Proper orientation of electrophile toward nucleophilic residue
* Feasible reaction geometry (distance, angle)
* Avoidance of nonspecific reactivity as covalent binders have a tendency to have high non-specific binding to other molecules in the body, causing a higher potential of toxicity.
What you prioritize
* Geometry over docking score
* Accessibility and selectivity of the reactive residue
*Integration note:* Protein–Ligand MD can later validate pre-reactive stability before covalent bond formation.
#### **D. Protein–Protein Interaction (PPI) Modulators**
Primary engines
* **Docking** (large, shallow interfaces)
* **BoltzGen Co-Folding**
* **Pharmacophore Analysis**
**Key focus**
* Hotspot engagement, targeting key residues at the PPI interface that contribute the most to binding free energy
* Surface complementarity ensuring that you have geometric and electrostatic fit between the ligands and the protein interface surface
* Disruption or stabilization of interface contacts where the compound could either disrupt the native protein protein contacts, or stabilize them like a molecular glue between proteins.
**What you prioritize**
* Shape complementarity along the interface
* Multi-point interaction patterns, ensuring engagement across the interfaces
* Interface-specific binding rather than engagements in other regions of the protein
4. **Interaction Pattern Analysis**
After screening, abstract away from individual poses. Use **Pharmacophore Analysis** to:
* Identify interaction motifs shared by top hits
* Confirm alignment with desired mechanism
* Filter out compounds that bind “correctly” but not *usefully*
This step ensures hits are mechanistically meaningful, not just high-scoring. The Pharmacophore Engine also will enable you to iterate on key motifs on the molecule with functional groups to optimize binding to key residues in the pocket.
5. **Dynamic or Energetic Validation (Optional)**
For high-confidence or high-cost decisions, escalate selectively. Depending on modality:
* **Protein–Ligand MD** to validate pose stability and conformational effects along with a breakdown of the energies using snapshot Free Energy calculations
* **Ligand–Membrane MD** for permeability- or membrane-driven mechanisms for protein targets on membrane interfaces
* **ABFE / RBFE** to refine ranking among closely related candidates within narrower chemical spaces.
This step is not required for all campaigns but strengthens confidence where needed.
## **Results**
* A modality-specific hit list aligned with your biological objective
* Structural explanations for *how* compounds engage the target
* Reduced false positives from generic screening
* Clear mechanistic rationale for experimental follow-up
## **Now What?** *I have hits matched to my desired mechanism, but what’s next?*
Typical next steps include:
* Focused experimental validation
* Mechanism-specific hit expansion
* Multi-parameter optimization
* Transition into lead optimization workflows
## **Why Revilico?**
Revilico enables mechanism-aware hit identification by allowing users to:
* Explicitly encode intent (inhibitor vs activator vs covalent, etc.)
* Configure screening and analysis engines accordingly
* Preserve structural interpretability throughout the workflow
* Seamlessly escalate into deeper validation or optimization pipelines
This ensures that hits are not just binders, but binders that behave the way you need them to.
At hit identification, not all hits are equal. Different discovery goals require fundamentally different binding behaviors, and a generic virtual screen often fails to distinguish them. Common challenges include:
* Docking scores that don’t reflect functional outcome (inhibition vs activation)
* Difficulty identifying covalent or mechanism-based binders
* Binding poses that look plausible but don’t explain functional modulation
* Mixing multiple binding modalities in a single hit list
This workflow is designed to explicitly tailor hit identification to your intended mechanism of action, producing hits that are aligned with *how* you want the target to be modulated.
## **Solution**
This workflow uses Revilico’s binding chemistry engines to customize hit discovery based on binding modality, combining structural filtering, interaction analysis, and (when needed) dynamic or energetic validation. The shared core workflow is: Target Preparation → Mechanism-Aware Docking → Interaction & Motif Analysis → Hit Prioritization.
Where the workflow diverges is in:
* How docking is configured
* What interactions are emphasized
* Which downstream validation steps are applied
Integration with MD, FEP, QSAR, and generative chemistry is possible once modality-specific hits are identified, and the campaign has reached an iterative optimization stage.
## **What Data Do I Need to Provide?**
Required
* Target protein structure (experimental or predicted)
* Compound library (or libraries) to screen
Recommended
* Known ligands or reference compounds for the desired modality
* Information about active sites, allosteric sites, or reactive residues
Optional
* Cofactors, ions, or partner proteins
* Experimental benchmarks for validation
## **Workflow**
1. **Define the Desired Binding Modality**
Before screening, explicitly define *what kind of hit you want*.\
Common criteria include:
* Inhibitors (competitive, non-competitive)
* Activators / positive allosteric modulators
* Covalent binders
* Allosteric binders
* Stabilizers / disruptors of protein–protein interactions
* Membrane-active or permeability-driven binders (when relevant)
This decision informs every downstream configuration choice to ensure that binding and engagement are translating to biologically relevant functions.
2. **Prepare Target Structures Based on Modality**
Different modalities require different structural contexts.
* Active-site binders (inhibitors):
* Prepare catalytic or orthosteric site
* Use known substrate or inhibitor coordinates when available as a reference
* Allosteric modulators / activators:
* Identify non-canonical pockets via literature, pocket detection, or prior MD
* Multiple conformations may be needed
* Covalent binders:
* Identify reactive residues (e.g., Cys, Ser, Lys)
* Ensure correct protonation and residue accessibility
Primary engines used
* **Docking** (for pose hypothesis)
* Optional **Protein Water MD** (to reveal transient or cryptic pockets) with potential follow up by the Pocket Search Engine to identify pockets of interest.
This step will give you one or more docking-ready target representations aligned with the mechanism.
3. **Mechanism-Aware Virtual Screening**
#### **A. Inhibitor Discovery**
Primary engines
* **Static Docking** → **Flexible Docking** (for refinement)
Key focus
* Competitive occupation of known binding pocket
* Strong, well-oriented interactions with catalytic residues
* Consistent low-energy poses
What you prioritize
* Affinity trends, especially hits that are reading out lower/negative kcal/mol energies
* Pose stability, looking at physically feasible binding and lack of steric clashes
* Overlap with known inhibitor binding modes, attaching to key amino acids of interest in the pocket
#### **B. Activators / Allosteric Modulators**
Primary engines
* **Docking** (alternative pockets)
* **Pharmacophore Analysis**
* **Molecular Dynamics Simulation Protein Ligand and Protein Water for Calibrations**
Key focus
* Binding outside the active site
* Interactions that stabilize specific protein conformations
* Poses that plausibly alter protein dynamics, and can be measured through molecular dynamics and Root Mean Squared Fluctuation (RMSF) values
What you prioritize
* Pocket specificity, ensuring that you are engaging with the right allosteric pocket
* Interaction networks rather than raw affinity scores, and physically feasible conformations
* Consistency across protein conformations
Validating the results
* In order to validate that your designed compound is an allosteric modulator, you can put the pose into molecular dynamics simulation over longer time span and look for RMSF value increases across longer time scales, in the active site of the given protein.
* Your designed compound should reveal a larger fluctuation pattern in the protein after ligand engagement compared to just protein in water MD
#### **C. Covalent Binder Identification**
Primary engines
* **Docking** (pose feasibility)
* **Pharmacophore Analysis** (reactive geometry validation)
Key focus
* Proper orientation of electrophile toward nucleophilic residue
* Feasible reaction geometry (distance, angle)
* Avoidance of nonspecific reactivity as covalent binders have a tendency to have high non-specific binding to other molecules in the body, causing a higher potential of toxicity.
What you prioritize
* Geometry over docking score
* Accessibility and selectivity of the reactive residue
*Integration note:* Protein–Ligand MD can later validate pre-reactive stability before covalent bond formation.
#### **D. Protein–Protein Interaction (PPI) Modulators**
Primary engines
* **Docking** (large, shallow interfaces)
* **BoltzGen Co-Folding**
* **Pharmacophore Analysis**
**Key focus**
* Hotspot engagement, targeting key residues at the PPI interface that contribute the most to binding free energy
* Surface complementarity ensuring that you have geometric and electrostatic fit between the ligands and the protein interface surface
* Disruption or stabilization of interface contacts where the compound could either disrupt the native protein protein contacts, or stabilize them like a molecular glue between proteins.
**What you prioritize**
* Shape complementarity along the interface
* Multi-point interaction patterns, ensuring engagement across the interfaces
* Interface-specific binding rather than engagements in other regions of the protein
4. **Interaction Pattern Analysis**
After screening, abstract away from individual poses. Use **Pharmacophore Analysis** to:
* Identify interaction motifs shared by top hits
* Confirm alignment with desired mechanism
* Filter out compounds that bind “correctly” but not *usefully*
This step ensures hits are mechanistically meaningful, not just high-scoring. The Pharmacophore Engine also will enable you to iterate on key motifs on the molecule with functional groups to optimize binding to key residues in the pocket.
5. **Dynamic or Energetic Validation (Optional)**
For high-confidence or high-cost decisions, escalate selectively. Depending on modality:
* **Protein–Ligand MD** to validate pose stability and conformational effects along with a breakdown of the energies using snapshot Free Energy calculations
* **Ligand–Membrane MD** for permeability- or membrane-driven mechanisms for protein targets on membrane interfaces
* **ABFE / RBFE** to refine ranking among closely related candidates within narrower chemical spaces.
This step is not required for all campaigns but strengthens confidence where needed.
## **Results**
* A modality-specific hit list aligned with your biological objective
* Structural explanations for *how* compounds engage the target
* Reduced false positives from generic screening
* Clear mechanistic rationale for experimental follow-up
## **Now What?** *I have hits matched to my desired mechanism, but what’s next?*
Typical next steps include:
* Focused experimental validation
* Mechanism-specific hit expansion
* Multi-parameter optimization
* Transition into lead optimization workflows
## **Why Revilico?**
Revilico enables mechanism-aware hit identification by allowing users to:
* Explicitly encode intent (inhibitor vs activator vs covalent, etc.)
* Configure screening and analysis engines accordingly
* Preserve structural interpretability throughout the workflow
* Seamlessly escalate into deeper validation or optimization pipelines
This ensures that hits are not just binders, but binders that behave the way you need them to.