> ## Documentation Index
> Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
> Use this file to discover all available pages before exploring further.
# Screening Compound Libraries
> Prioritize a Compound Library Against a Target Using Virtual Screening
## **The Problem You Are Trying to Solve**
*“I have a biological target and a large compound library that would be expensive and time-consuming to screen experimentally. I want to computationally prioritize a smaller, higher-confidence subset of compounds before committing to HTS.”*
 At the hit identification stage, experimental HTS can be:
* Costly and slow at large library scales
* Noisy, with high false-positive and false-negative rates
* Difficult to iterate on quickly
This workflow is designed to front-load computational triage, allowing you to focus experimental resources on compounds most likely to bind and be biologically relevant.
## **Solution**
This workflow uses Revilico’s Virtual Screening and Binding Chemistry engines to down-select large libraries into a ranked, mechanistically interpretable shortlist.
The primary prioritization chain is: Target Preparation → Virtual Screening (Docking) → Pose & Score Analysis → Optional Refinement (Flexible / Ensemble Docking).
This approach allows users to:
* Rapidly screen large libraries
* Eliminate obvious non-binders
* Preserve structural insight into *why* compounds were prioritized using structure activity relationships and chemical space analysis.
* Seamlessly escalate promising candidates into deeper simulations if needed
Other engines (QSAR, MD, FEP, generative chemistry) can be layered on later as confidence or optimization needs increase.
## **What Data Do I Need to Provide?**
Required
* Target protein structure (experimental or predicted)
* Compound library (CSV with SMILES strings), or if you’d like access to our partner’s 2M liquid stock libraries for direct delivery after computational screening, reach out to us.
Recommended
* Known binding site or reference ligand (to define docking region)
* Any known cofactors, ions, or biologically relevant states of the target
Optional
* Multiple protein conformations (for flexible or ensemble docking)
* Experimental benchmark compounds (for calibration and validation).This usually will consist of ligand sets with corresponding experimentally determined activity values.
## **Workflow**
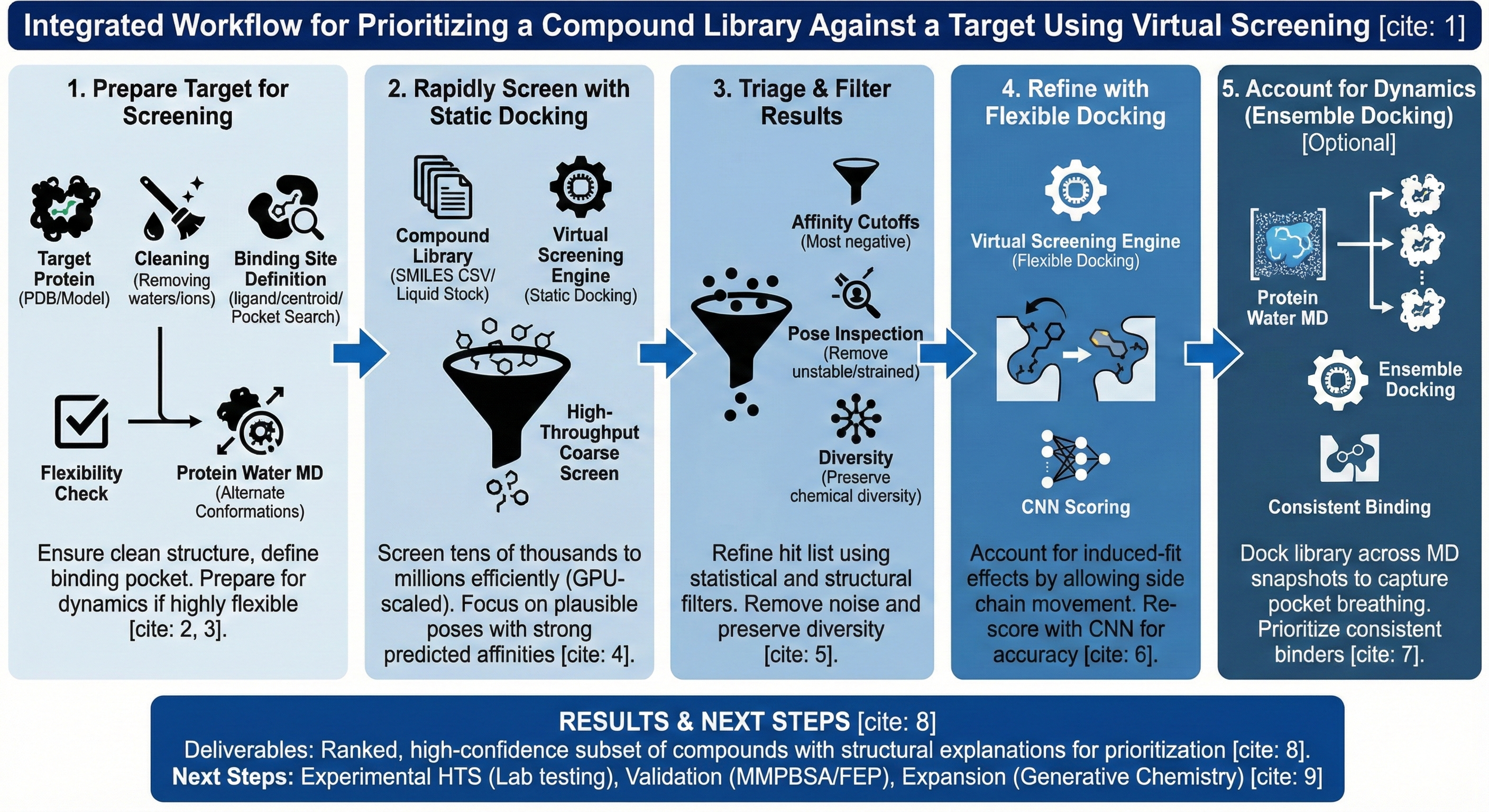
1. **Prepare the Target for Screening**
Before screening, ensure the target structure is suitable for docking. On Revilico, users typically:
* Upload or retrieve a protein structure (PDB or predicted model)
* Clean the structure (remove waters, ions, irrelevant ligands)
* Define the binding site (known ligand coordinates, residue-based centroid, or pocket detection)
If target flexibility or uncertainty is expected, this step can later integrate **Protein Water MD** to generate alternate conformations, and to assess for certain vulnerabilities static algorithms may possess for certain protein target systems..
This results in a docking-ready protein structure with a defined binding region. If you are still assessing the proper protein pocket, you can utilize our pocket search engine after generating structures using AlphaFold, Boltz2, or OpenFold.
2. **Rapidly Screen the Full Library with Static Docking**
Begin with high-throughput prioritization. This step prioritizes speed and coverage, not perfect accuracy.
Use **Static Docking** to:
* Screen tens of thousands to millions of compounds efficiently using GPU scaled docking algorithms
* Predict binding poses and approximate affinities
* Quickly eliminate compounds with poor shape or interaction complementarity
What you’re looking for
* Strong predicted affinities relative to the bulk library
* Plausible poses that occupy the intended binding pocket
* Consistency across multiple poses
This gives a ranked list of compounds with predicted binding scores and poses. After the initial screening utilizing static docking, the library will still have uncertainty in the predicted binding affinities as these algorithm types tend to have higher rates of false positives and negatives, necessitating a deeper screen with the following algorithms listed below.
3. **Triage and Filter Docking Results**
Refine the initial hit list using structural and statistical filters. This step ensures that prioritization is not driven by docking noise alone.
Users typically:
* Apply affinity cutoffs. The more negative the activity, the better the target engagement.
* Remove compounds with unstable or highly strained poses
* Inspect pose clustering to avoid single-pose artifacts
* Preserve chemical diversity while downselecting
This filtering step outputs a reduced, higher-quality candidate set suitable for deeper analysis.
4. **Refine Binding Predictions with Flexible Docking**
For the most promising compounds, increase physical realism by utilizing the flexible docking engine which allows the protein side chain to be flexible on certain selected residues.
Use **Flexible Docking** to:
* Allow selected protein side chains to move
* Capture induced-fit effects missed by rigid docking
* Re-rank compounds based on improved pose accuracy
* Re-score poses generated with convolutional neural network (CNN) filters, to get better pose accuracies
This step is especially valuable when:
* The binding site is flexible, and critical amino acids for binding are known
* Small chemical differences need better resolution to differentiate activity cliffs within narrower chemical spaces
This results in a refined ranking with higher confidence in binding modes.
5. **Account for Protein Dynamics with Ensemble Docking (Optional, but Highly Recommended)**
If the target is highly dynamic or known to adopt multiple binding-competent states:
* Use **Protein Water MD** to generate conformational snapshots, and to get refined parameters quantifying the protein’s behaviors in different solvents and time scales.
* Apply **Ensemble Docking** across these structures to assess target engagement over the course of the protein’s trajectory in solution.
This captures:
* Pocket breathing
* Transient sub-pockets
* Conformational selection effects of ligand engagement
This will result in prioritized compounds that bind consistently across protein states.
## **Results**
* A ranked, prioritized compound subset suitable for experimental testing
* Structural explanations for prioritization decisions
* Reduced HTS cost and time by focusing on high-value candidates
* Clear upgrade path into hit validation and optimization workflows
## **Now What?** *I have a prioritized list, but what’s next?*
Common next steps include:
* Experimental HTS or focused biochemical assays
* Binding mechanism analysis (MD, pharmacophore analysis)
* Hit expansion or optimization using generative chemistry
* Energetic validation with MMPBSA or FEP for top candidates
## **Why Revilico?**
Revilico enables cost-effective hit identification by combining:
* High-throughput **Virtual Screening**
* Physically grounded refinement (**Flexible / Ensemble Docking**)
* Transparent structural and energetic interpretation
* Seamless escalation into deeper simulation or optimization workflows
This allows teams to screen smarter, not bigger, dramatically reducing experimental burden while increasing the likelihood of meaningful hits. Like all other engines, properly calibrating your screens with some sort of experimental data subset will increase the interpretability of the results and allow for more accurate filtering criterias.
At the hit identification stage, experimental HTS can be:
* Costly and slow at large library scales
* Noisy, with high false-positive and false-negative rates
* Difficult to iterate on quickly
This workflow is designed to front-load computational triage, allowing you to focus experimental resources on compounds most likely to bind and be biologically relevant.
## **Solution**
This workflow uses Revilico’s Virtual Screening and Binding Chemistry engines to down-select large libraries into a ranked, mechanistically interpretable shortlist.
The primary prioritization chain is: Target Preparation → Virtual Screening (Docking) → Pose & Score Analysis → Optional Refinement (Flexible / Ensemble Docking).
This approach allows users to:
* Rapidly screen large libraries
* Eliminate obvious non-binders
* Preserve structural insight into *why* compounds were prioritized using structure activity relationships and chemical space analysis.
* Seamlessly escalate promising candidates into deeper simulations if needed
Other engines (QSAR, MD, FEP, generative chemistry) can be layered on later as confidence or optimization needs increase.
## **What Data Do I Need to Provide?**
Required
* Target protein structure (experimental or predicted)
* Compound library (CSV with SMILES strings), or if you’d like access to our partner’s 2M liquid stock libraries for direct delivery after computational screening, reach out to us.
Recommended
* Known binding site or reference ligand (to define docking region)
* Any known cofactors, ions, or biologically relevant states of the target
Optional
* Multiple protein conformations (for flexible or ensemble docking)
* Experimental benchmark compounds (for calibration and validation).This usually will consist of ligand sets with corresponding experimentally determined activity values.
## **Workflow**
1. **Prepare the Target for Screening**
Before screening, ensure the target structure is suitable for docking. On Revilico, users typically:
* Upload or retrieve a protein structure (PDB or predicted model)
* Clean the structure (remove waters, ions, irrelevant ligands)
* Define the binding site (known ligand coordinates, residue-based centroid, or pocket detection)
If target flexibility or uncertainty is expected, this step can later integrate **Protein Water MD** to generate alternate conformations, and to assess for certain vulnerabilities static algorithms may possess for certain protein target systems..
This results in a docking-ready protein structure with a defined binding region. If you are still assessing the proper protein pocket, you can utilize our pocket search engine after generating structures using AlphaFold, Boltz2, or OpenFold.
2. **Rapidly Screen the Full Library with Static Docking**
Begin with high-throughput prioritization. This step prioritizes speed and coverage, not perfect accuracy.
Use **Static Docking** to:
* Screen tens of thousands to millions of compounds efficiently using GPU scaled docking algorithms
* Predict binding poses and approximate affinities
* Quickly eliminate compounds with poor shape or interaction complementarity
What you’re looking for
* Strong predicted affinities relative to the bulk library
* Plausible poses that occupy the intended binding pocket
* Consistency across multiple poses
This gives a ranked list of compounds with predicted binding scores and poses. After the initial screening utilizing static docking, the library will still have uncertainty in the predicted binding affinities as these algorithm types tend to have higher rates of false positives and negatives, necessitating a deeper screen with the following algorithms listed below.
3. **Triage and Filter Docking Results**
Refine the initial hit list using structural and statistical filters. This step ensures that prioritization is not driven by docking noise alone.
Users typically:
* Apply affinity cutoffs. The more negative the activity, the better the target engagement.
* Remove compounds with unstable or highly strained poses
* Inspect pose clustering to avoid single-pose artifacts
* Preserve chemical diversity while downselecting
This filtering step outputs a reduced, higher-quality candidate set suitable for deeper analysis.
4. **Refine Binding Predictions with Flexible Docking**
For the most promising compounds, increase physical realism by utilizing the flexible docking engine which allows the protein side chain to be flexible on certain selected residues.
Use **Flexible Docking** to:
* Allow selected protein side chains to move
* Capture induced-fit effects missed by rigid docking
* Re-rank compounds based on improved pose accuracy
* Re-score poses generated with convolutional neural network (CNN) filters, to get better pose accuracies
This step is especially valuable when:
* The binding site is flexible, and critical amino acids for binding are known
* Small chemical differences need better resolution to differentiate activity cliffs within narrower chemical spaces
This results in a refined ranking with higher confidence in binding modes.
5. **Account for Protein Dynamics with Ensemble Docking (Optional, but Highly Recommended)**
If the target is highly dynamic or known to adopt multiple binding-competent states:
* Use **Protein Water MD** to generate conformational snapshots, and to get refined parameters quantifying the protein’s behaviors in different solvents and time scales.
* Apply **Ensemble Docking** across these structures to assess target engagement over the course of the protein’s trajectory in solution.
This captures:
* Pocket breathing
* Transient sub-pockets
* Conformational selection effects of ligand engagement
This will result in prioritized compounds that bind consistently across protein states.
## **Results**
* A ranked, prioritized compound subset suitable for experimental testing
* Structural explanations for prioritization decisions
* Reduced HTS cost and time by focusing on high-value candidates
* Clear upgrade path into hit validation and optimization workflows
## **Now What?** *I have a prioritized list, but what’s next?*
Common next steps include:
* Experimental HTS or focused biochemical assays
* Binding mechanism analysis (MD, pharmacophore analysis)
* Hit expansion or optimization using generative chemistry
* Energetic validation with MMPBSA or FEP for top candidates
## **Why Revilico?**
Revilico enables cost-effective hit identification by combining:
* High-throughput **Virtual Screening**
* Physically grounded refinement (**Flexible / Ensemble Docking**)
* Transparent structural and energetic interpretation
* Seamless escalation into deeper simulation or optimization workflows
This allows teams to screen smarter, not bigger, dramatically reducing experimental burden while increasing the likelihood of meaningful hits. Like all other engines, properly calibrating your screens with some sort of experimental data subset will increase the interpretability of the results and allow for more accurate filtering criterias.