Why Use This Engine?
In the documentation below, we will use Revilico’s RevFEP engine to compute binding free energies between protein targets and small molecule ligands using alchemical free energy perturbation (FEP). RevFEP automates the full pipeline from raw protein-ligand complex PDB files through structure preprocessing, alchemical network construction, GPU-accelerated simulation, and statistical analysis to produce DDG or DG estimates with uncertainty quantification. It is the highest-accuracy binding affinity method available on the platform and is the appropriate engine when docking scores are insufficient to distinguish closely related lead compounds.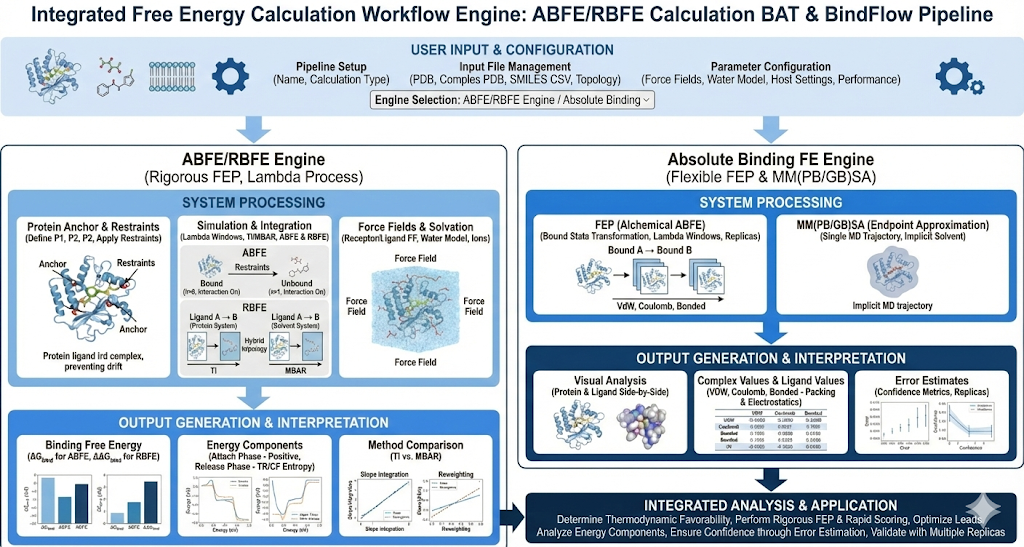
Background
Free energy perturbation is a rigorous thermodynamics-based method for computing binding affinity differences between related molecules. Rather than using empirical scoring functions, FEP simulates the alchemical transformation between two chemical states using molecular dynamics and computes the free energy difference from the work distribution of the transformation. When applied to a series of ligands against a protein target, FEP produces binding affinity rankings that are far more accurate than docking or MM-GBSA, with mean absolute errors typically below 1 kcal/mol for well-prepared systems. RevFEP is built on OpenFE 1.10 and OpenMM, deployed on AWS Batch for scalable GPU execution. It supports four calculation types covering the range from rapid relative ranking to absolute binding free energy estimation, and implements seven advanced protocol stages including non-equilibrium MD, REST2 enhanced sampling, adaptive lambda optimization, and MM-GBSA pre-filtering.Calculation Types
RBFE (Relative Binding Free Energy) RBFE calculates the relative binding affinity between pairs of ligands (DDG_bind). It is the most efficient engine for lead optimization where a series of structural analogues must be ranked against a known binder. Pairs of ligands are connected in a perturbation network, and for each pair a single alchemical transformation is simulated. The OpenFE HREX/MBAR protocol is used with Hamiltonian replica exchange across lambda windows. ABFE (Absolute Binding Free Energy) ABFE calculates the absolute binding free energy of a single ligand to a protein (DG_bind) by running two independent simulation legs: the complex leg (protein and ligand together) and the solvent leg (ligand in water only). ABFE is slower than RBFE but provides absolute DG values without requiring a reference compound. SepTop (Separated Topologies) SepTop uses separated topology alchemical transformations where the two end-state ligands are represented as independent topology files simultaneously scaled between lambda = 0 and lambda = 1. This allows perturbations between structurally dissimilar ligands where a maximum common substructure mapping would be too small for reliable RBFE. AHFE (Absolute Hydration Free Energy) AHFE calculates the free energy of transferring a ligand from vacuum into water (DG_hydration). It is a protein-free calculation used to validate force field parameters and assess ligand solvation characteristics before running more expensive binding calculations.Structure Preparation Pipeline
Protein Preparation The input protein-ligand complex PDB is parsed to separate protein ATOM records from ligand HETATM records. The protein cleaning pipeline then runs in sequence: standard residues are retained while non-standard residues and crystallographic artifacts are handled; missing heavy atoms are rebuilt using PDBFixer; missing hydrogen atoms are added at pH 7.0; disulfide bonds and CONECT records are preserved; and the cleaned structure is written aspreprocessed/protein.pdb for use as the ProteinComponent in all subsequent calculations.
Ligand Extraction and Processing
Ligands are extracted from the input PDB using a two-tier strategy that handles both GNINA docking output (HETATM ligand with CONECT bond order records) and generic docking output (ATOM ligand with UNL residue name). The extracted ligand is passed through RDKit for hydrogen addition, 3D coordinate assignment, and SMILES round-trip validation to ensure chemical correctness.
3D Alignment for RBFE and SepTop
For multi-ligand campaigns, ligands extracted from independent docking runs may be oriented differently in the binding pocket. The engine performs Maximum Common Substructure (MCS) alignment using RDKit rdFMCS with the first ligand as the reference, applying rdMolAlign.AlignMol() with the MCS atom map for 3D superposition. Ligands with an MCS smaller than 3 atoms are retained with a warning but are not aligned.
Partial Charge Assignment
Partial charges are pre-assigned during preprocessing using the selected small molecule force field (OpenFF Sage or Rosemary by default, GAFF2 available as an alternative). Pre-assigning charges avoids running antechamber at simulation time and ensures consistent charge treatment across all ligands in the campaign.
Perturbation Network
For RBFE and SepTop, ligands are connected in a perturbation network that defines which pairs will be simulated. The network topology determines the total number of alchemical transformations and therefore the size of the AWS Batch array job. The engine constructs the network using Kartograf atom mapping, which relies on 3D spatial overlap of aligned ligand poses to identify the maximum common substructure for each pair.Three-Phase AWS Pipeline
Phase 1: Setup All inputs are preprocessed, the perturbation network is constructed, and OpenFE transformation JSON objects are written toopenfe_setup/transformations/. A manifest file lists all transformations with their array indices for the run phase.
Phase 2: Run (GPU Array Job)
Each AWS Batch array element runs one alchemical transformation. The transformation is loaded from its JSON, the lambda schedule is applied, and HREX molecular dynamics is executed across all lambda windows on a GPU. Each window produces a dhdl.xvg file. The GPU array job runs all transformations in parallel, making the total wall time proportional to the slowest individual transformation rather than the sum.
Phase 3: Gather and Analysis
After all array elements complete, the gather phase collects per-transformation result JSONs and runs MBAR (Multistate Bennett Acceptance Ratio) analysis via alchemlyb to produce the final DG or DDG estimates with uncertainty. Results are written to openfe_summary/summary.tsv and analysis_output/ containing MBAR convergence plots and the DDG matrix.
Advanced Protocol Stages
Stage 1: Advanced Force Fields The protein force field can be set to AMBER ff14SB (default) or ff19SB. The small molecule force field can be set to OpenFF Sage 2.2 (default), OpenFF Rosemary, or GAFF2. Water model options include TIP3P, TIP4P-Ew, and SPC/E. Stage 2: Non-Equilibrium MD (NE-MD) NE-MD implements the Crooks Fluctuation Theorem using the Bennett Acceptance Ratio estimator for rapid DG estimation. Rather than running long equilibrium simulations at multiple lambda windows, NE-MD switches the Hamiltonian rapidly between two end states and measures the forward and reverse work distributions. BAR analysis of the bidirectional work values yields a free energy estimate: Where is the Fermi function and the averages are over forward and reverse work trajectories. Stage 3: REST2 Enhanced Sampling Replica Exchange with Solute Tempering version 2 (REST2) improves conformational sampling by selectively scaling the ligand (solute) interaction energies across replicas at effective temperatures higher than the target. This allows the ligand to overcome torsional barriers that would not be crossed in standard equilibrium MD, improving convergence without the cost of scaling the full system. Stage 4: Adaptive Lambda Windows After a pilot FEP run, this stage analyzes the MBAR phase-space overlap between adjacent lambda windows. Windows with overlap below the target threshold are identified, and additional intermediate windows are suggested. The refined lambda schedule is written toadaptive_lambda/lambda_suggestions.json for use in the production run.
Stage 5: Force Field Torsion Scan
Dihedral energy scans are run across all rotatable bonds in each ligand using multiple force field methods (OpenFF, GAFF2, and semiempirical xTB). Comparing energy profiles across methods identifies potential force field parameter quality issues before committing to expensive FEP simulations.
Stage 6: Tighter ABFE Settings
Increases the lambda window count and tightens the restraint configuration for ABFE calculations, improving accuracy at the cost of longer simulation time. Recommended for final-stage ABFE calculations on clinical candidates.
Stage 7: MM-GBSA Pre-Filter
Before committing to FEP, all ligands are rapidly scored against the protein using MM-GBSA with Generalized Born (OBC2) implicit solvent:
All three energies use the same OBC2 solvation model (solvent dielectric 78.5, solute dielectric 1.0) with local minimization and short equilibration, so systematic errors cancel in the difference. Ligands below the score cutoff are pruned from the perturbation network, reducing the total number of FEP transformations and the associated compute cost.
Running the Engine
Inputs
| Parameter | Default | Description |
|---|---|---|
| Protein-ligand complex PDB | Required | One PDB per ligand (RBFE/SepTop) or single PDB (ABFE/AHFE) |
| Calculation type | rbfe | rbfe, abfe, septop, or ahfe |
| Protein force field | ff14SB | ff14SB or ff19SB |
| Small molecule force field | OpenFF Sage 2.2 | openff-2.2, openff-rosemary, or gaff2 |
| Water model | TIP3P | tip3p, tip4pew, or spce |
| Lambda windows | 11 | Number of alchemical intermediate windows |
| Advanced stages | None | Any combination of the 7 enhancement stages |
| MM-GBSA cutoff | -20 kcal/mol | Score threshold for pre-filter pruning (Stage 7) |
Outputs
- DDG matrix: Relative binding free energy differences between all ligand pairs (RBFE/SepTop)
- DG table: Absolute binding free energies per ligand (ABFE/AHFE)
- Uncertainty estimates: MBAR standard error per transformation
- MBAR convergence plots: Phase-space overlap matrices per transformation
- Perturbation network visualization: Graph of all simulated ligand pairs
- MM-GBSA scores: Pre-filter scores and pruned network (Stage 7)
- Adaptive lambda report: Suggested refined lambda schedule (Stage 4)
- NE-MD work distributions: Forward and reverse work histograms with BAR estimates (Stage 2)
- Torsion scan profiles: Per-bond dihedral energy plots across force fields (Stage 5)