Documentation Index
Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
Use this file to discover all available pages before exploring further.
Why Use this product?
ADMET-AI rapidly predicts 41 pharmacokinetic and toxicity properties, including absorption, distribution, metabolism, excretion, and toxicity endpoints, using machine learning models trained on Therapeutics Data Commons datasets. This enables drug discovery teams to filter thousands of compounds from virtual screening or generative AI in minutes, eliminating molecules with poor drug-like properties (low solubility, hERG liability, hepatotoxicity) before expensive synthesis and testing. By providing fast, accurate ADMET predictions with the highest performance on industry benchmarks, ADMET-AI bridges the gap between computational hit identification and experimental validation, dramatically reducing the cost and time of lead optimization for later stage tasks after activity optimizations.
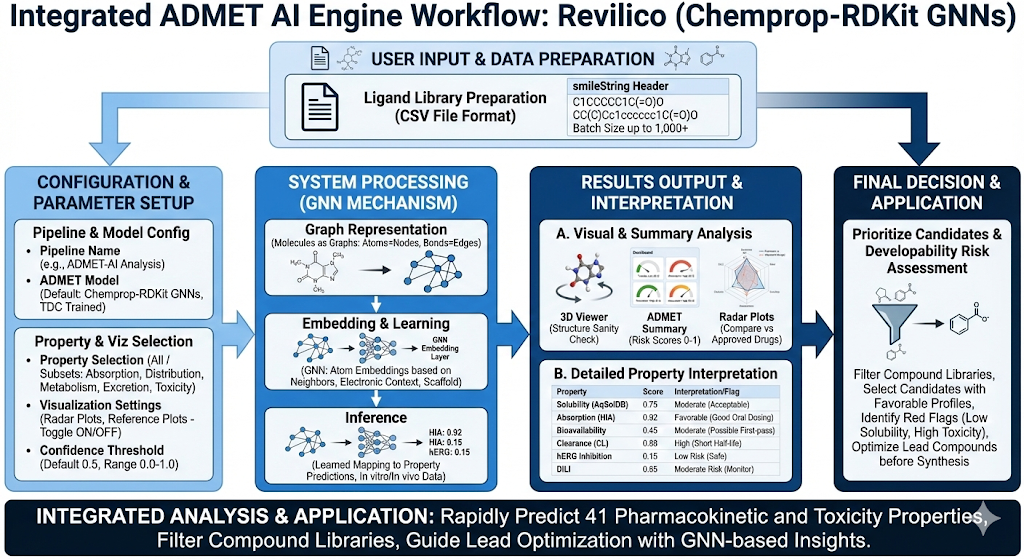
Background
Whether the ligand will engage with and bind to the protein is only one part of the question when it comes to whether a drug can be deemed effective and be passed into the clinic. Predicting the compound properties answers the other part of the question. It answers whether the drug will deliver the proper effect while maintaining other important properties and minimizing toxicity. By creating a high throughput screening system that can predict the properties of a specific compound, chemists will be able to prioritize molecules with a higher chance of succeeding in development.
Introducing Revilico’s ADMET AI engine, it is a data driven machine learning model, trained on large experimental ADMET datasets that is designed to rapidly assess developability risks of small molecules solely based on their chemical structure, enabling users to assess compounds based on absorption, metabolism, distribution, excretion, and/or toxicity before synthesis and experimental testing.
Now how does it work? This model is based on a principle that many ADMET outcomes are strongly correlated with high-level chemical structure and physicochemical properties rather than detailed molecular mechanisms (i.e. Membrane permeability correlates with size and polarity). Rather than calculating these properties using extensive experimentation, ADMET-AI deploys a machine learning model that can learn these properties from the data. First we begin with our user input, or SMILES strings. We then ask the question, how can we transform this string into something the model can understand and extract value from. We understand the principles that the behavior of an atom depends on its neighbor, functional groups behave differently depending on scaffold context, and small structural changes can lead to large biological effects. Using this principle of the relationships between atoms and the graph like nature of a molecule on a 2D/3D coordinate system, we can deploy a Graph Neural Network (GNN), where the model learns an atomistic representation of its local chemical environment including nearby functional groups, electronic context, and structural constraints imposed by the scaffold. Here we can produce a value or embedding in which the model can understand.
The model itself was trained on ADMET datasets that contain a wide range of small molecules with known outcomes measured from in vitro assays, in vivo studies, and biochemical screens. During training, the model learns a mapping from molecular embeddings to ADMET properties. At inference time, the input molecule’s embedding is passed through this learned mapping to generate predictions.
Interactive Results Viewer
Explore ADMET AI prediction results interactively. Select a molecule to view its predicted properties across absorption, distribution, metabolism, excretion, and toxicity categories.