Documentation Index
Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
Use this file to discover all available pages before exploring further.
Why Use this product?
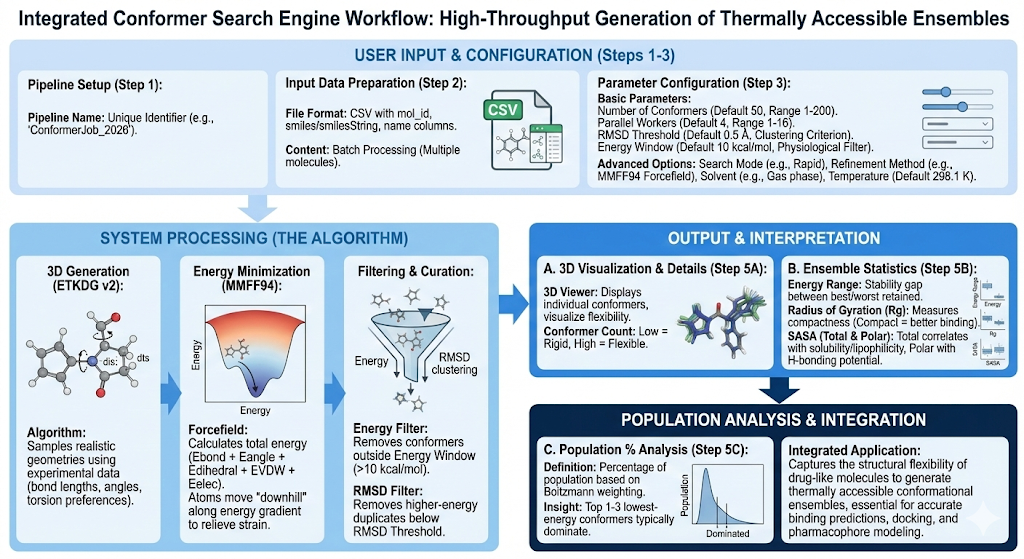
The Conformer search engine is a computational tool that delivers thermally accessible conformation ensembles that capture the structural flexibility of drug-like molecules essential for accurately binding predictions and structure activity relationship analysis. You will use this engine when you need to generate comprehensive ensembles of low-energy 3D molecular conformations for drug discovery applications like molecular docking or pharmacophore modeling. You can utilize this engine to analyze intrinsic compound conformational energies when cross referencing generated/docking ligand protein poses. Large deviations in conformation needs to be analyzed to ensure favorable energetic compositions and changes.
Background
Conformation refers to the specific 3D shape a molecule can adopt. We may have a single molecule, however this molecule can adopt several different plausible shapes. But why is this important? The conformation of a molecule affects how effectively it binds to targets, influencing potency, absorption, and side effects. Because conformations have different energies, finding lower energy conformations allows researchers to better predict drug-target interactions, and design molecules for optimal binding. When discovering different conformations, chemists often discover them using advanced spectroscopy, X-ray crystallography, and electron microscopy. This can be considered a very time consuming and expensive task. This is where Revilico’s Conformer Search Engine comes into place. This engine operates as a high throughput conformer predictor that can both effectively and speedily discover many different conformations of your small molecule, saving both the time and money to run any experimentation.
Now how does it work? We first start with our SMILES strings. We can convert this into a 3D structure Before introducing the ETKDG v2 algorithm. This algorithm was designed based on experimental conformational data where statistics regarding typical bond lengths, typical angles, and preferred torsion angles were hard coded into this algorithm. This algorithm will take the base molecule and generate different possible confirmations ensuring that bonds and non-bonded atoms fall within a realistic distance range, certain rotations are sampled more frequently than others, and rings are constrained to known geometries. The step by step process of generating a single conformer is first to build a distance bounds matrix, followed by sampling torsion angles based on experimental data, solving the geometry problem ensuring that atoms are placed in a manner that satisfies the constraints, and finally the algorithm checks for any possible steric clashes. We will now generate a large panel of different conformations for each molecule that was inputted to get a probability landscape of that conformer’s existence.
We now introduce the MMFF94 forcefield. In this step we will compute the force-field energy, slightly move the atoms to create an energetic downhill motion towards an energetic minimum, and stop when the forces are small, relieving strain, fixing small clashes and producing a local minimum conformer. This forcefield is based on these equations that assess internal compound interactions:
E=∑interactions
Etotal=Ebonds+Eangle+Edihedral+EVDW+Eelec
Ebond=∑kb(r−r0)2
Eangle=∑k0(θ−θ0)2
Edihedral=∑2Vn(1+cos(nϕ−γ))
EVDW=∑4ϵ[(rσ)12−(rσ)6]
Eelec=∑(qi×qj)/(4πϵ0rij)
This equation of total energy will calculate the total energy results derived from the use of the forcefield. When we want to move the atoms downhill to get to a local minimum conformer, this will be based on calculating the gradient of the forcefield/ energetic landscape as denoted by:
F=−∇E
Where ∇E calculates the gradient derivate of the energy in each possible direction, in our case along the x, y and z plane. This can be denoted as
∇E(r)=(∂x∂E,∂y∂E,∂z∂E)
We will do this for each conformer that was generated to produce a list of different conformers and their associated energies. To curate the panel of conformers we will return, we will first filter all conformers based on their energy, with lowest energy being highly prioritized and highest energy being listed last. We will then down-select our panel based on our energy window. Our energy window is the energy difference between the target conformer generated and the conformer with the lowest energy. This can be denoted as a
∇E=Econformer−Emin
Where ∇E must be less than the size of the energy window. If it does not satisfy this requirement we will get rid of the conformer from #down-selected list. The next step will be to filter based on the Root Mean Square Deviation (RMSD) thresholds, which ensures proper geometric feasibility and accuracy. For this we do not want conformers that are too similar to each other, or in our case less than the RMSD difference threshold measured between each conformer generated. What we will do is evaluate each pair of conformers, removing the conformer with the higher energy. In the end we will only return conformers that are greater than our RMSD threshold (to ensure conformer diversity), and that perform better, with lower calculated energies.
Interactive Results Viewer
Explore conformer search results interactively. View 3D conformer structures with an energy slider, Boltzmann populations, and shape descriptors.