Documentation Index
Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
Use this file to discover all available pages before exploring further.
Why Use this product?
The Retrosynthesis engine is an AI powered tool that generates complete multi-step synthesis pathways, identifies required building blocks, and provides alternative routes ranked by feasibility enabling rapid route planning, synthesizability assessment, and identification of chemical analogs utilizing retrosynthetic analysis. This engine is best used when you have a target molecule and need to determine how to synthesize it from commercially available starting materials.
Background
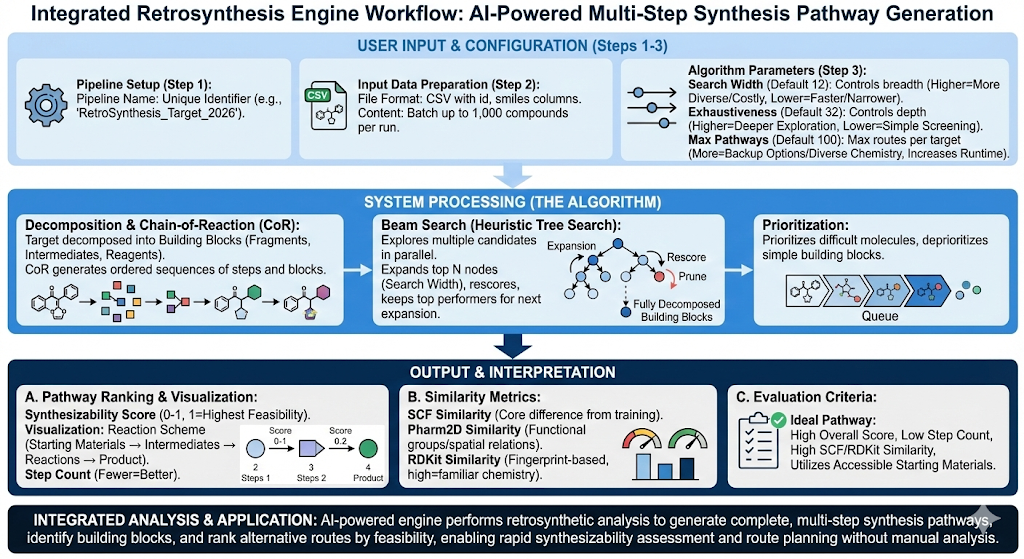
Synthesizability is a key metric in evaluating whether a molecule can become a drug in practice. Without synthesizability, even the most potent, selective, or computationally promising molecule cannot become practically made in the laboratory. In a laboratory, molecules are synthesized through a stepwise chemical synthesis starting from existing well characterized compounds that are drawn from commercial reagent catalogs, fragment libraries, and previously synthesized intermediates. Essentially, this compound library will be our building blocks for each of our molecules, and will be utilized in several reaction steps. When understanding synthesisability, we are asking the question of whether our molecule be designed using the current compound library that we have on hand? This is where Revilico’s Retrosynthesis Engine comes into place to help plan, organize, and arrange logistics for challenging synthesis steps of downstream lead series acquisition and testing. This engine has the ability to test this parameter and generate hypotheses for chemists from a computational standpoint, and in a high throughput manner, to enable chemists to thoroughly screen their library of lead molecules for synthesizability, saving both time and money.
Diving into the workflow of how this engine works, we realize that this is an advanced tree search problem, or in other words we are trying to find all possible combinations of building blocks needed to synthesize this molecule. We will first start with our molecule of interest and try to decompose it into the different building blocks (i.e. fragments, intermediates, and reagents) that can be curated into our target molecule of interest that needs to eventually be tested in the lab.
After designing a lead molecule computationally, the largest question will be how this compound is segmented into smaller substituents that can be readily accessible to chemists in the lab.Traditionally, this process would be handled primarily through the chemists experiences and theoretical recollection, but now we can begin to determine the building block combinations required to build this molecule. This model has been trained on experimental reaction data from a large library of known compounds including, commercially available reagents, common intermediates, and frequently used medicinal chemistry fragments where the input is reactant molecules and the output is the observed product. This relationship can be encoded by the following equation
(Reactant Set)+(Reaction)→(Product)
From the data the model will learn which reactions are chemically valid, which functional groups participate in which transformation, which precursor-reaction combinations are plausible,what the reaction conditions are, and how likely a given transformation is based on the precedent reaction sets encoded within the model.
Based on this foundation, we can dive deeper into Chain-of-Reaction (CoR) reasoning. Rather than predicting a single step, the model will generate CoR sequences where a CoR is an ordered sequence of building blocks and reaction steps. When executed in the forward direction, the chain will reconstruct the target molecule using prior knowledge of reactants and reaction sets within its training set. We can think of the entire Chain of Reaction sequence as a recipe, where each node will represent the progress in the recipe. We can think of each node as the step needed to create the final product, and with this model architecture, we are able to fragment the features that are fed in to represent a cohesive retrosynthesis problem. For example, one step is how we make the dough. Water, yeast, and flour are the reactants, mixing is the reaction and dough is the product. The CoR architectures are able to fully resolve these steps with different confidences to paint a picture of experimental protocols that should be taken to get to target products with on hand reactants.
Now we introduce the core algorithm. Our algorithm is based on a tree search, specifically tailored towards defining retrosynthesis as a decomposition problem. If we think about a retrosynthetic pathway as a tree/network with nodes and edges, we have defined the maximum number of child nodes that can be allocated per parent node, where a set of child nodes are the decomposition steps of the parent node. Nodes at the same hierarchy level represent different possible configurations to create the same parent node for a given task. We have also determined the depth of the ‘tree’ or network, which signifies how many times an ingredient can be decomposed. For example, if we are looking at baking bread, a depth of 3 could start with bread, decompose to dough, then decompose the dough into water, yeast, and flour, and finally decompose the flour into wheat.
We can now introduce the Beam Search algorithm. The Beam Search algorithm is a heuristic tree-search algorithm that explores many candidate solutions in parallel while strictly limiting how many are actively pursued at each step. In other words at each step we have a set of nodes and we will only take a few of the top performing ones and expand those. For example, say we have a search width of 3. First we start with our parent node and generate the top 3 children nodes. The next step will be expanding out each of these 3 nodes, so at the next level we will have 9 total nodes. At this step we will rescore and only take the top 3 at the same level again, then expand those, repeating the same steps until we hit our max depth. So essentially we start at 1 node, expand to 3, then expand to 9 only keeping the top 3 performing ones, then expanding to 9 again keeping the top 3, repeating over and over until we reach our depth length. We are searching for the best possible configurations or components that contribute to the decompositions.
Now how do we decompose each node? Each node is a set of different reactants and reactions. For now we will look at the reactants. In our bread analogy, say our dough is the current parent node. Then the children nodes or reactants will be our water, yeast and flour. The algorithm will look at each of these molecules or reactants in our set (water, yeast, and flour), then evaluate for the most difficult or unresolved molecule set for expansion. Molecules already in the building-block are deprioritized. So in our case the next level will be expanding flour and keeping water and yeast the same since those are building blocks, things that are elementary enough and do not need to be decomposed further. We can then decompose flour into grains. Now at this new node our reactants will be water, yeast, and grains.
In the end we should have a set or sets of reactants from our building-block library that can be used to synthesize our molecule. For the purpose of this algorithm, we utilize commercially available reactant sets primarily to ensure logistically smooth syntheses.
Interactive Results Viewer
Explore retrosynthesis results interactively. View synthesis pathways ranked by feasibility score, reaction steps, similarity metrics, and 3D molecular structures.