Why Use this product?Documentation Index
Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
Use this file to discover all available pages before exploring further.
Revilico’s Boltz Co-Folding engine is an AI powered tool that goes beyond structure prediction alone, also predicting binding affinity of protein-ligand or protein-protein complexes simultaneously. This engine enables rapid virtual screening, hit discovery, and ligand optimization for early-stage drug discovery without requiring expensive molecular dynamics simulations.
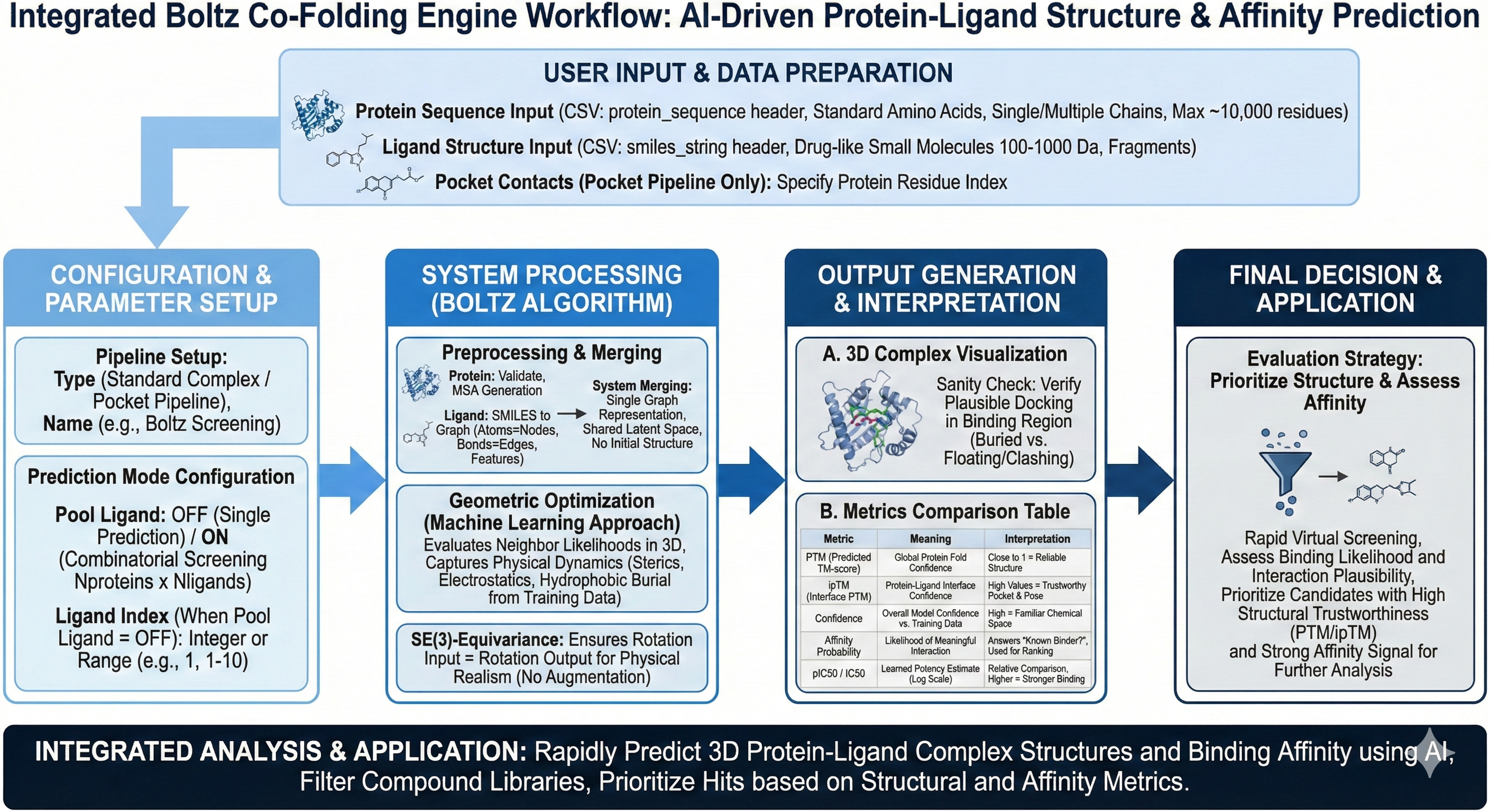
For most typical workflows, we would have to run our Alphafold Engine to generate our 3D protein structure, then use our pocket search engine to determine the pocket in which we want to bind to the ligand, then run our docking engine in order to find the binding affinity metrics as well as the best pose for the small molecules we would like to test. This is a multistep process that can both take a significant amount of time and compute. This is where Revilico’s Boltz-Cofolding engine comes into place. This engine’s main goal is to provide fast, structure-level hypotheses for where a ligand is likely to bind, how the protein may fold or rearrange around the ligand, and assess whether the interaction looks plausible compared to other known protein-ligand complexes. If our goal is to quickly screen a protein and a set of ligands to see whether they should be sent into the wet lab, we can use Boltz Cofolding to confirm whether it has favorable conformations and activity.
What is Boltz? Boltz is a machine learning based co-folding model trained on known protein ligand complex structures and functional activity scores. It will predict a single protein ligand complex geometry, confidence metrics for fold and interface, and provides the user a learned affinity signal. Now we can dive into how this engine worksFirst we upload our Protein Sequences and our ligand SMILES strings. Our next step would be individual preprocessing of the protein sequences and the ligand SMILES strings, similar to how we operate the AlphaFold engine. Simply put, we will first validate the sequence (i.e. check that all amino acids are valid, assign residue indices and handle chain breaks if multiple proteins are present). We then undergo a Multiple Sequence Alignment (MSA) integration where MSA helps to align the sequence and structures to similar proteins, derived from evolutionary conservation of structure, allowing the model to see which residues have mutational and evolutionary conservation over time. To read more about how MSA works please refer to the Protein Folding Documentation here. Now for preprocessing the ligands, the SMILES strings are then converted to a graph structure where atoms are the nodes and bonds are the edges and each node is assigned features (i.e. element type, formal charge, hybridization, and aromaticity. These descriptors and graph representations of the ligands are then sent into an AI model to help predict the rest of the downstream outputs. The next step would be merging the protein and ligand into a single system placing the protein residues and ligand atoms in the same graph representation, where there is no structure, only identities and relationships of the different input parameters. The job of the model is to then figure out how these different features can be represented together on a shared latent space (i.e. a representation of the multi-variable system with a lower dimension representation of vectors). Boltz algorithm takes the approach of learning the conserved underlying patterns of physics and chemistry that exist within the data rather than solving mathematical equations for binding interactions and energies at each step. Simply put, this means that the model will capture physics dynamics such as steric repulsion, favorable electrostatics, hydrophobic burial, and entropic preferences based on the experimental data that the model was trained on rather than having to calculate these dynamics at each step. At this point within the algorithm, we have our network arrays of protein residues and ligand atoms representations/features. We will undergo a process of geometric optimization and iteration until we have reached our final optimized conformational state. The nodes in the graph each have their own properties (i.e. charge, hybridization, etc). The model will look at neighbors of atoms (nodes as represented by the algorithm), and evaluate how likely these nodes should be near each other within a specified conformation in a 3D predicted structure. Based on this likelihood, the nodes will update their internal representation accordingly and reevaluate. As the algorithm progresses and begins maximization of physical feasibility of certain conformations, interactions will increase their likelihood of being physically represented properly until the best conformation is submitted. Lastly it is important to note that Boltz is an SE(3)-equivariant which means that when we rotate or translate the input, the output rotates or translates in the same way (i.e. the model does not align with absolute protein structure orientations, only relative geometry matters within the system. This is critical for physical realism and avoids any need for data augmentation as it guarantees that bond lengths stay consistent, angles behave properly, and structures obey spatial laws, all derived through MSA templates as a starting point.