Documentation Index
Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
Use this file to discover all available pages before exploring further.
Why Use this product?
Revilico’s Custom Model Training Engine is a transfer learning tool that applies pre-trained models to your curated compound library creating custom models that generate chemistry aligned with your organization’s structural preferences. IP landscape, and SAR knowledge for downstream library generation and lead optimization campaigns. This engine is best used when you need to fine-tune the De Novo Library Generation Engine or the Molecular Optimization engine, enabling the generation of molecules that statistically resemble your training dataset rather than generic drug-like compounds.This feature helps you to establish your ‘chemical prior’ model that can be used for molecular optimization, allowing you to sample from the chemical space of your input molecular library and surrounding spaces.
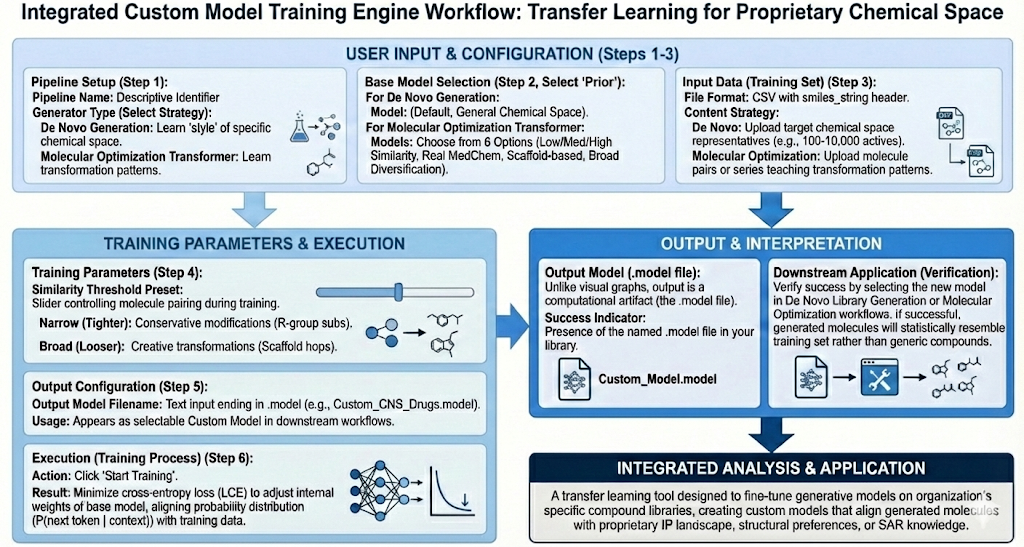
Background
When running the De Novo Library Generation Engine and the Molecular Optimization engine, the basis for generation of our datasets is the prior model which encapsulates a large chemical space. However, if our task requires us to only generate molecules within a certain chemical space. We will not be able to use the reinvent prior model, and we will now need a model that is only trained on that specific chemical space we are interested in. This is where Revilico’s Custom Model Training Engine comes into place. The goal of this engine is to take a compound library and transform it into a model that can be used downstream in De Novo Library Generation or in Molecular Optimization.
How does it work? We have two types of library generators. With the De Novo Generation generator, what we will do is input a compound library, and the output will be molecules that are similar in that chemical space. With Molecular Optimization Transformer, we will input a compound library, typically one that has a core scaffold with slight differences or medicinal chemistry transformations. This model will look at the transformation of the molecules within the library, focused on generating chemically plausible analogs that reflect the learned transformation patterns within that lead series or compound set.
With both of these models, it generally operates by first starting with our start token or some fragment of our SMILES string. The job of the model is to predict the next token given the previous tokens. It can be denoted by this probability.
P(next token∣previous token,context)
With De Novo Generation it can be more precisely defined as:
P(next token∣previous token)
Where the statistics come from all molecules in the training set. With Molecular Optimization it can be defined as:
P(output token∣input molecule tokens,previous output tokens)
Which means it is conditioned on the specific input molecule, answering the question, given this molecule, how do chemists usually modify it in this project, through learned associations of the chemical space.
During training the model is corrected in this process where the model’s prediction is compared to the actual next token in your dataset. If it guesses wrong, its internal weights are adjusted slightly. This will use cross-entropy loss as the loss function where the goal of the model is to minimize the cross entropy loss. Cross entropy loss can be denoted by the following formula:
LCE=−i∑yilog(pi)
Where i is the index token y is the true label as denoted by 1 or 0 and p is the predicted probability for token i.
We will end up with a model with updated model weights in accordance to the input training data. This model will now generate new models from scratch with either goal in mind of creating molecules that resemble a particular set or analog molecules similar to our training data set.