Documentation Index
Fetch the complete documentation index at: https://docs.revilico.bio/llms.txt
Use this file to discover all available pages before exploring further.
Why Use this product?
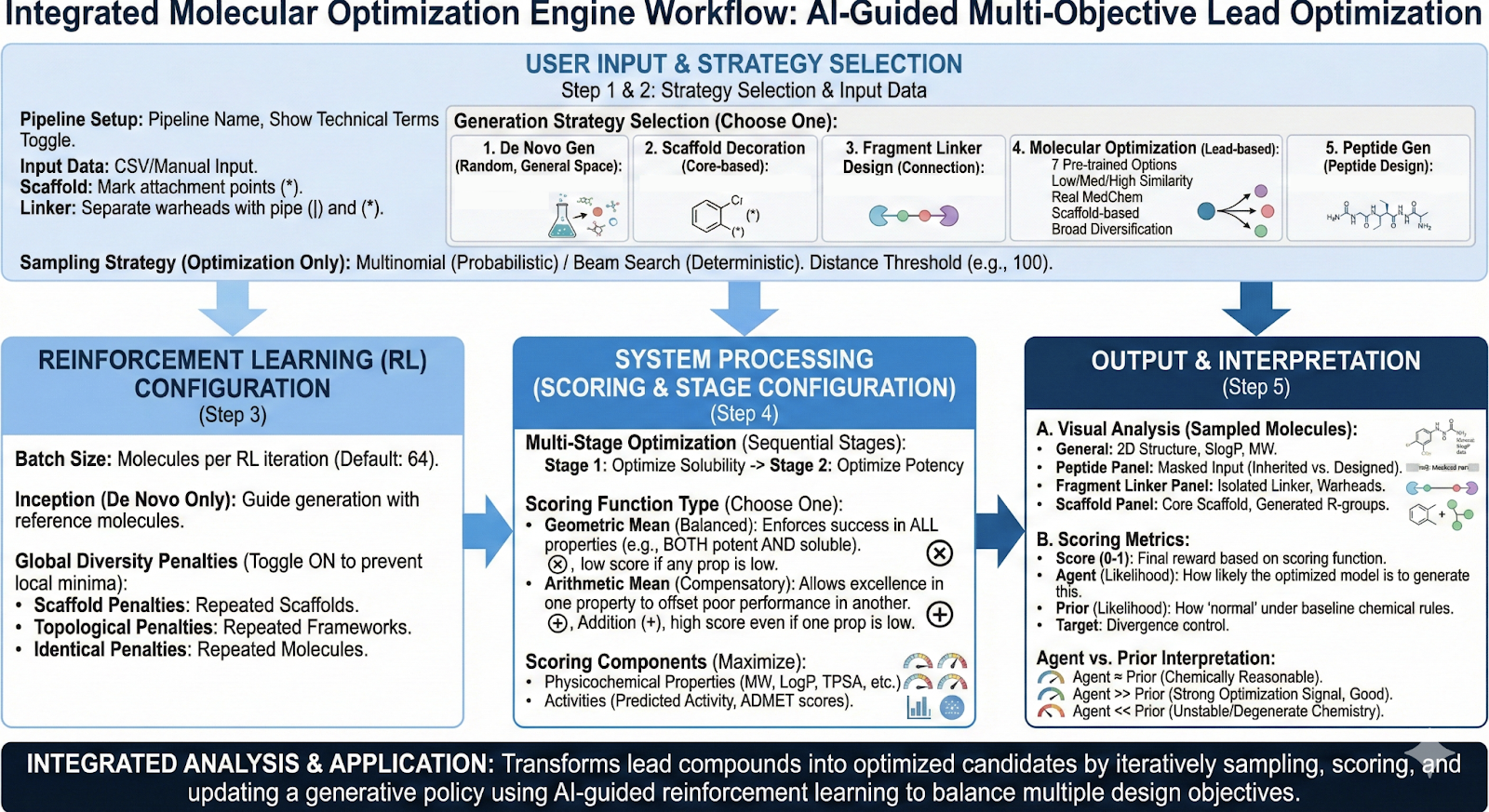
Revilico’s Molecular Optimization engine aims to generate a compound library from a starting molecule that is optimized for the properties that the user prioritizes. This engine will transform lead compounds into optimized drug candidates through AI guided reinforcement learning that simultaneously balances multiple design objectives, potency selectivity, ADME properties, and synthesizability. For Multi-parameter optimization, reinforcement learning serves as a great algorithmic solution to optimizing leads for several different properties simultaneously.
Background
Revilico’s Molecular Optimization Engine has relatively the same backbone as the De Novo Library Generation engine, where it is based on a chemical language model where SMILES strings are sentences, and each token is a part of a SMILES string and is a certain word. To recap how this model works is that we first have a start token, this is a small fragment of our SMILES string. Using this
P(next token∣previous tokens)
We will construct the probability distribution of the next token or next part of the SMILES string based on the current string we have and the objectives we are trying to solve for across each generative step. We will then sample from the distribution and append it to the previous tokens and repeat this process until we reach our end token which is optimized towards a specific objective, scored by several engines that assess physicochemical properties, activity, or other criterion.
Now what is the difference between De Novo Library Generation and Molecular Optimization? With Molecular Optimization we will need to define a scoring objective (i.e. what is one property we would like to optimize for), then we will need to reweight this objective in comparison to the other metrics in which the model is capturing. Essentially De Novo generation is just brainstorming the different molecules we can generate based off of a starting protein target, or just randomly generated molecules with certain optimized properties. With Optimization, we are sampling, scoring, then updating the generator so it will produce better molecules in line with our scoring objective, across each iteration of reinforcement learning.
Now let us dive into the full workflow. First our scoring function can be defined as:
Score(molecule)→x∈[0,1]
here X is some sort of scaled value from 0 (poor) to 1 (excellent). Things that can go into a score can be physiochemical property targets (e.g. MW in a range, LogP in a range, TPSA), similarity constraints using fingerprint similarity, penalties and hard constraints (e.g. remove toxic substructures, reactive groups), activity calculations using Revilico’s FEP suite, or other binding affinity scoring functions on the platform, and external predictors (e.g. ADMET AI outputs or solubility predictors).
Before diving into the algorithm, we need to understand these two pieces of terminology: prior and policy.The prior is the baseline generative model trained to reproduce chemically valid molecules with an unbiased distribution while the policy is the optimized version of the prior, fine-tuned towards specific objectives (biased distributions). The prior is the assumptions of the model’s parameters before seeing any input data or scoring objections. The policy is a map of the actions we should take which will lead to maximizing long term rewards(in terms of more optimized molecules).
We first start with our first De Novo Library Generation step. Here we can say that our policy = prior (i.e. we do not know anything about the data and therefore there are no biases in the model). Here we will generate our first set of molecules then score them. When we score we will be able to update our policy, by updating rewards and penalties for the steps based on whether it will generate preferred or undesirable molecules. As we go through more iterations within the reinforcement learning process, the policy will tighten depending on the scoring function, helping the generator to shift the prior towards a more biased chemical space, optimized for your specific properties. By assigning rewards and penalties we will have a token probability bias. To ensure that we do not keep doing the same path over and over and keep ourselves in a local maxima (the max reward of what we have seen so far, but not the max reward of the entire search space), we introduce a probability of exploring a different space rather than maximizing the total reward at that step to ensure molecular diversity. This will allow us to see if there is another path of generating a molecule that has a higher maximum reward than the current maximum. Note that it is important that we only do incremental changes during each iteration, allowing the model to learn which patterns actually improve performance over time. Essentially what we are doing is trying to balance exploitation of the reward with exploration of the search space (optimization of the given property while allowing the algorithm to have enough diverse exploration for novel hypotheses. We can define what optimization is trying to maximize with the following equation:
L(θ)=Ex∼πθ[R(x)−βKL(πθ(x)∥πprior(x))]
Where πθ(x) is the current policy, β controls how strongly the policy is anchored to the prior, with small β being aggressive optimization and large β being safe exploration, and KL being the regularization term that penalizes drifting too far from known chemistry.
Based on the score we generate we will be able to update our policy and repeat this process of optimization over and over again until we reach our panel of molecules.