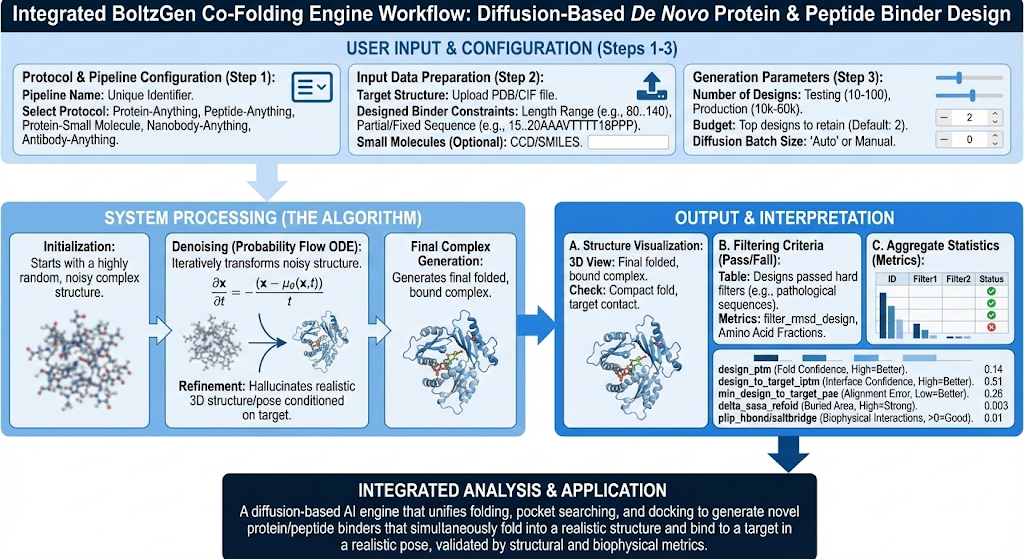
Why Use this product?
The BoltzGen Cofolding Engine is a diffusion based tool that generates thousands of candidate binders with validated binding poses enabling rapid discovery of high affinity binders for drug development, diagnostics, or protein engineering applications without requiring experimental screening. This tool is best used when you need to design novel protein or peptide binders against therapeutic targets through AI powered de novo generation that simultaneously optimizes binding affinity, structural stability, and sequence diversity.