The Problem You Are Trying to Solve
“I have a biological target and a large compound library that would be expensive and time-consuming to screen experimentally. I want to computationally prioritize a smaller, higher-confidence subset of compounds before committing to HTS.”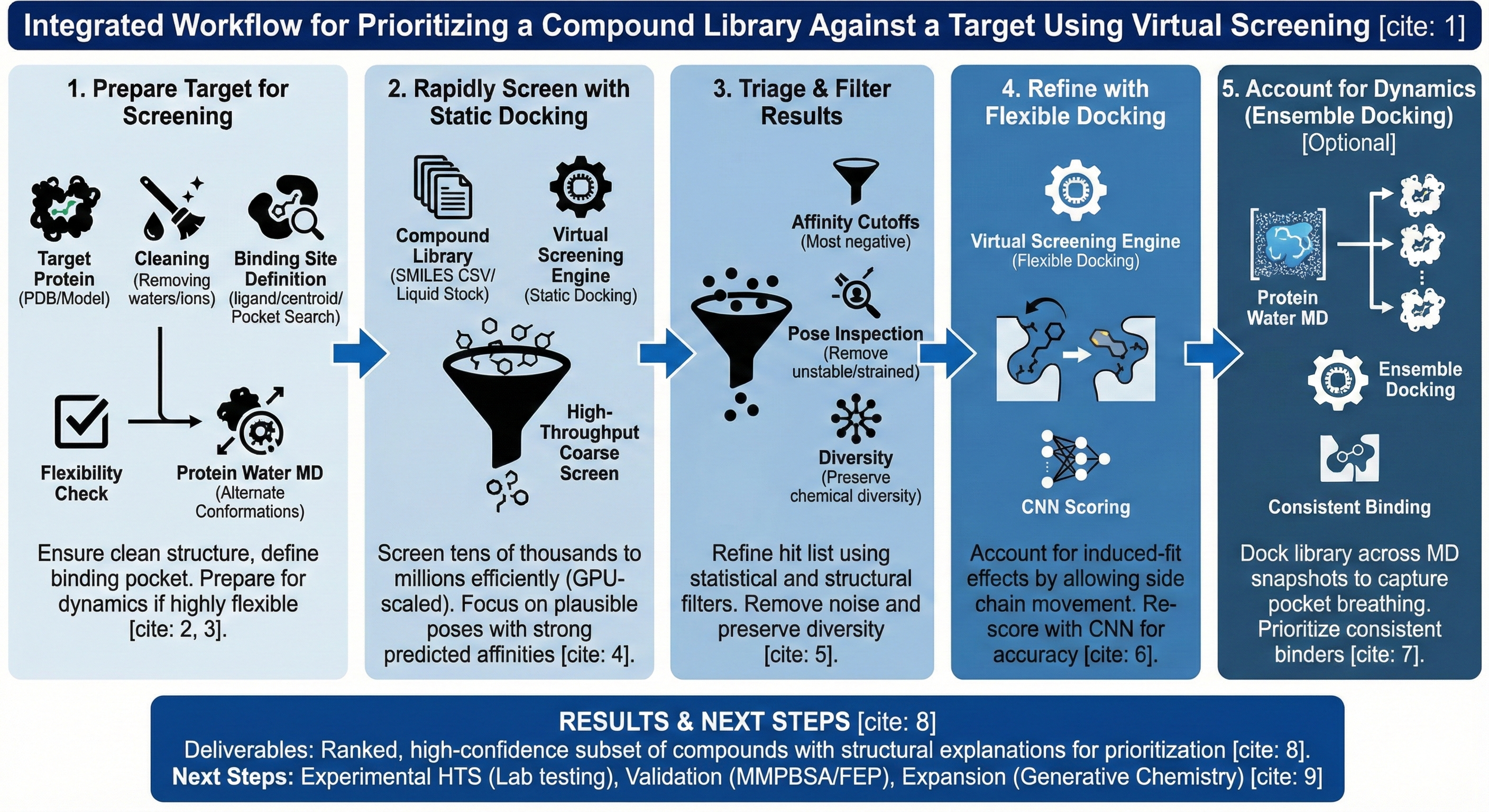
- Costly and slow at large library scales
- Noisy, with high false-positive and false-negative rates
- Difficult to iterate on quickly
Solution
This workflow uses Revilico’s Virtual Screening and Binding Chemistry engines to down-select large libraries into a ranked, mechanistically interpretable shortlist. The primary prioritization chain is: Target Preparation → Virtual Screening (Docking) → Pose & Score Analysis → Optional Refinement (Flexible / Ensemble Docking). This approach allows users to:- Rapidly screen large libraries
- Eliminate obvious non-binders
- Preserve structural insight into why compounds were prioritized using structure activity relationships and chemical space analysis.
- Seamlessly escalate promising candidates into deeper simulations if needed
What Data Do I Need to Provide?
Required- Target protein structure (experimental or predicted)
- Compound library (CSV with SMILES strings), or if you’d like access to our partner’s 2M liquid stock libraries for direct delivery after computational screening, reach out to us.
- Known binding site or reference ligand (to define docking region)
- Any known cofactors, ions, or biologically relevant states of the target
- Multiple protein conformations (for flexible or ensemble docking)
- Experimental benchmark compounds (for calibration and validation).This usually will consist of ligand sets with corresponding experimentally determined activity values.
Workflow
- Prepare the Target for Screening
- Upload or retrieve a protein structure (PDB or predicted model)
- Clean the structure (remove waters, ions, irrelevant ligands)
- Define the binding site (known ligand coordinates, residue-based centroid, or pocket detection)
- Rapidly Screen the Full Library with Static Docking
- Screen tens of thousands to millions of compounds efficiently using GPU scaled docking algorithms
- Predict binding poses and approximate affinities
- Quickly eliminate compounds with poor shape or interaction complementarity
- Strong predicted affinities relative to the bulk library
- Plausible poses that occupy the intended binding pocket
- Consistency across multiple poses
- Triage and Filter Docking Results
- Apply affinity cutoffs. The more negative the activity, the better the target engagement.
- Remove compounds with unstable or highly strained poses
- Inspect pose clustering to avoid single-pose artifacts
- Preserve chemical diversity while downselecting
- Refine Binding Predictions with Flexible Docking
- Allow selected protein side chains to move
- Capture induced-fit effects missed by rigid docking
- Re-rank compounds based on improved pose accuracy
- Re-score poses generated with convolutional neural network (CNN) filters, to get better pose accuracies
- The binding site is flexible, and critical amino acids for binding are known
- Small chemical differences need better resolution to differentiate activity cliffs within narrower chemical spaces
- Account for Protein Dynamics with Ensemble Docking (Optional, but Highly Recommended)
- Use Protein Water MD to generate conformational snapshots, and to get refined parameters quantifying the protein’s behaviors in different solvents and time scales.
- Apply Ensemble Docking across these structures to assess target engagement over the course of the protein’s trajectory in solution.
- Pocket breathing
- Transient sub-pockets
- Conformational selection effects of ligand engagement
Results
- A ranked, prioritized compound subset suitable for experimental testing
- Structural explanations for prioritization decisions
- Reduced HTS cost and time by focusing on high-value candidates
- Clear upgrade path into hit validation and optimization workflows
Now What? I have a prioritized list, but what’s next?
Common next steps include:- Experimental HTS or focused biochemical assays
- Binding mechanism analysis (MD, pharmacophore analysis)
- Hit expansion or optimization using generative chemistry
- Energetic validation with MMPBSA or FEP for top candidates
Why Revilico?
Revilico enables cost-effective hit identification by combining:- High-throughput Virtual Screening
- Physically grounded refinement (Flexible / Ensemble Docking)
- Transparent structural and energetic interpretation
- Seamless escalation into deeper simulation or optimization workflows