The Problem You Are Trying to Solve
“I have a binder to my protein, but I want to design synergistic or allosteric modulators to improve target engagement and reduce reliance on traditional, trial-and-error SAR campaigns.”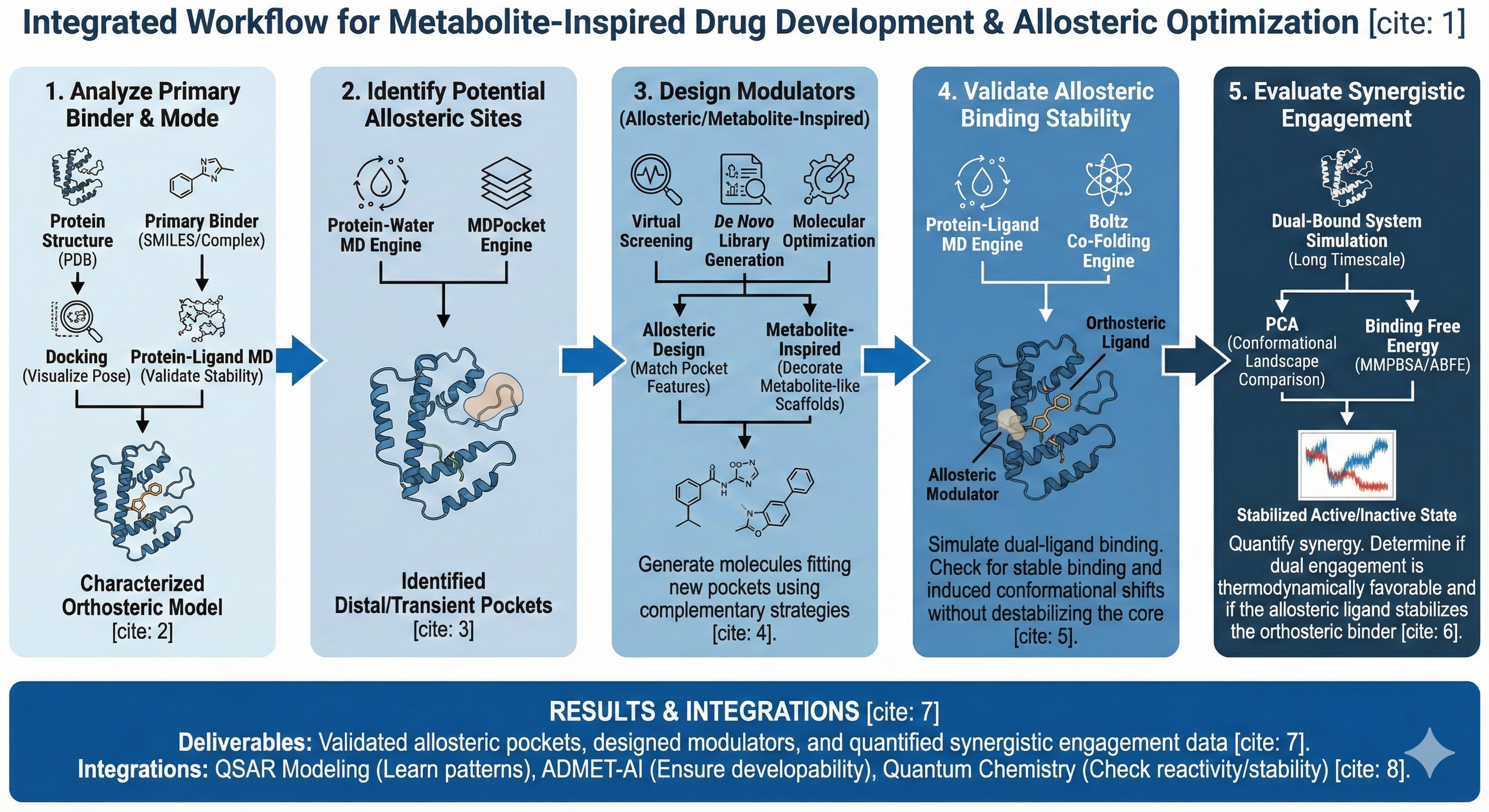
- Achieve desired potency in complex biological systems
- Improve selectivity over homologous proteins
- Modulate protein function dynamically (activation vs inhibition)
- Enhance stability or residence time
Solution
This workflow identifies potential allosteric sites, designs modulators inspired by other small molecule engagers like metabolites or secondary binding pockets, and computationally validates synergistic engagement before experimental investment.The primary design chain is: Primary Binder Analysis → Allosteric Site Identification → Allosteric Modulator Design → Binding & Stability Validation → Synergy Evaluation. This enables rational exploration of allosteric control and multi-site engagement with minimal experimental burden.
What Data Do I Need to Provide?
Required- Protein structure (PDB or predicted structure)
- Primary binder structure (SMILES or PDB complex)
- Known compound or metabolite structures (if metabolite-inspired design is desired)
- Known regulatory partners or secondary binders
- Any experimental binding/activity data for calibration
- Target conformational states of interest (active vs inactive)
- Known mutational or regulatory hotspot regions
Workflow
- Analyze the Primary Binder and Binding Mode
- Visualize the orthosteric binding pose with Docking
- Identify key residues and interaction motifs
- Validate stability and observe dynamic contacts with Protein-Ligand MD
- Determine which structural regions remain unoccupied
- Which residues drive binding?
- Are there flexible regions or distal pockets that move during MD?
- Does binding induce conformational shifts?
- Identify Potential Allosteric Sites
- Run Protein-Water MD to observe natural pocket breathing, and feed the simulations into MDPocket engine where transient pockets can be elucidated for further analysis.
- Analyze RMSF, SASA, and PCA outputs to identify flexible regulatory regions
- Examine transient pocket formation during MD trajectories
- Transient pockets forming distal to the active site
- Correlated motion between distant domains
- Stable but ligand-free cavities that open during simulation
- Design Allosteric or Metabolite-Inspired Modulators
- Run Virtual Screening / Docking on the allosteric pocket
- Use De novo Library Generation for new chemical matter
- Use Molecular Optimization to decorate metabolite-like scaffolds
- Use Pharmacophore Analysis to match pocket features
- Upload metabolite SMILES
- Identify shared motifs with endogenous ligands
- Preserve functional groups important for recognition
- An example of this would be kinase inhibitor designs replicating ATP structures that drive engagement for proteins that do downstream phosphorylation
- Validate Allosteric Binding Stability
- Initial assessments of the orthosteric ligand in the primary pocket can be done with docking, co-folding, or molecular dynamics simulations
- ProteinLigand MD for the allosteric binder alone
- Optionally simulate both orthosteric + allosteric ligand together. Revilico’s Boltz Co-Folding Engine allows for you to simulate multiple ligands binding within 1 structure, seeing how activity will change with just 1 ligand binding with the protein versus with both compounds.
- Using downstream Protein Ligand MD for either system will help elucidate pockets opening up on the protein with longer term dynamic time scales.
- Stable binding in the secondary pocket
- No destabilization of protein core
- Conformational shifts induced by allosteric binding
- Evaluate Synergistic Engagement
- Simulate dual-bound systems (orthosteric + allosteric ligand) with co-folding with pooled ligand pairs on one specific target and then running the ligands on Protein–Ligand MD simulations with long enough time spans to capture protein motion induced with ligands.
- Compare conformational landscapes via PCA
- Evaluate binding free energy shifts via:
- MMPBSA and MMGBSA (rapid estimate)
- ABFE/RBFE (higher rigor with alchemical transformations of the ligand)
- Does the allosteric ligand stabilize the orthosteric binder?
- Does the protein adopt a more favorable active/inactive conformation?
- Is dual engagement thermodynamically favorable?
Results
- Identified and validated allosteric pocket(s)
- Designed metabolite-inspired modulators
- Stability-confirmed binding models
- Quantified synergistic target engagement
- Reduced need for brute-force SAR experimentation
Integration with Other Engines (Optional)
This workflow can connect to:- QSAR Modeling (learn patterns from orthosteric + allosteric activity data)
- ADMET-AI (ensure new modulators remain developable)
- Quantum Chemistry (HOMO–LUMO, Geometry Optimization) for reactivity/stability checks
- Free Energy Perturbation for fine analog ranking
- scRNA-Seq Analysis to assess downstream pathway effects of modulation
Why Revilico?
Revilico enables metabolite-inspired and allosteric drug design by combining:- Structural insight (Docking + MD)
- Dynamic conformational analysis
- Generative chemistry capabilities
- Thermodynamic validation (FEP)
- Integrated interpretation tools