“I have a moderately active small molecule, and I want to improve it across multiple dimensions (potency, selectivity, developability, etc.) by exploring scaffold decoration and scaffold hopping, while preserving key motifs that drive favorable target interaction.”
- Multi-parameter goals often conflict (e.g., potency vs solubility vs lipophilicity) and optimizing structure for one property usually will do the opposite for other properties you are concerned with
- Scaffold hopping can accidentally remove the very interaction features responsible for binding
- Minor changes can unexpectedly shift binding mode, strain, or developability risk
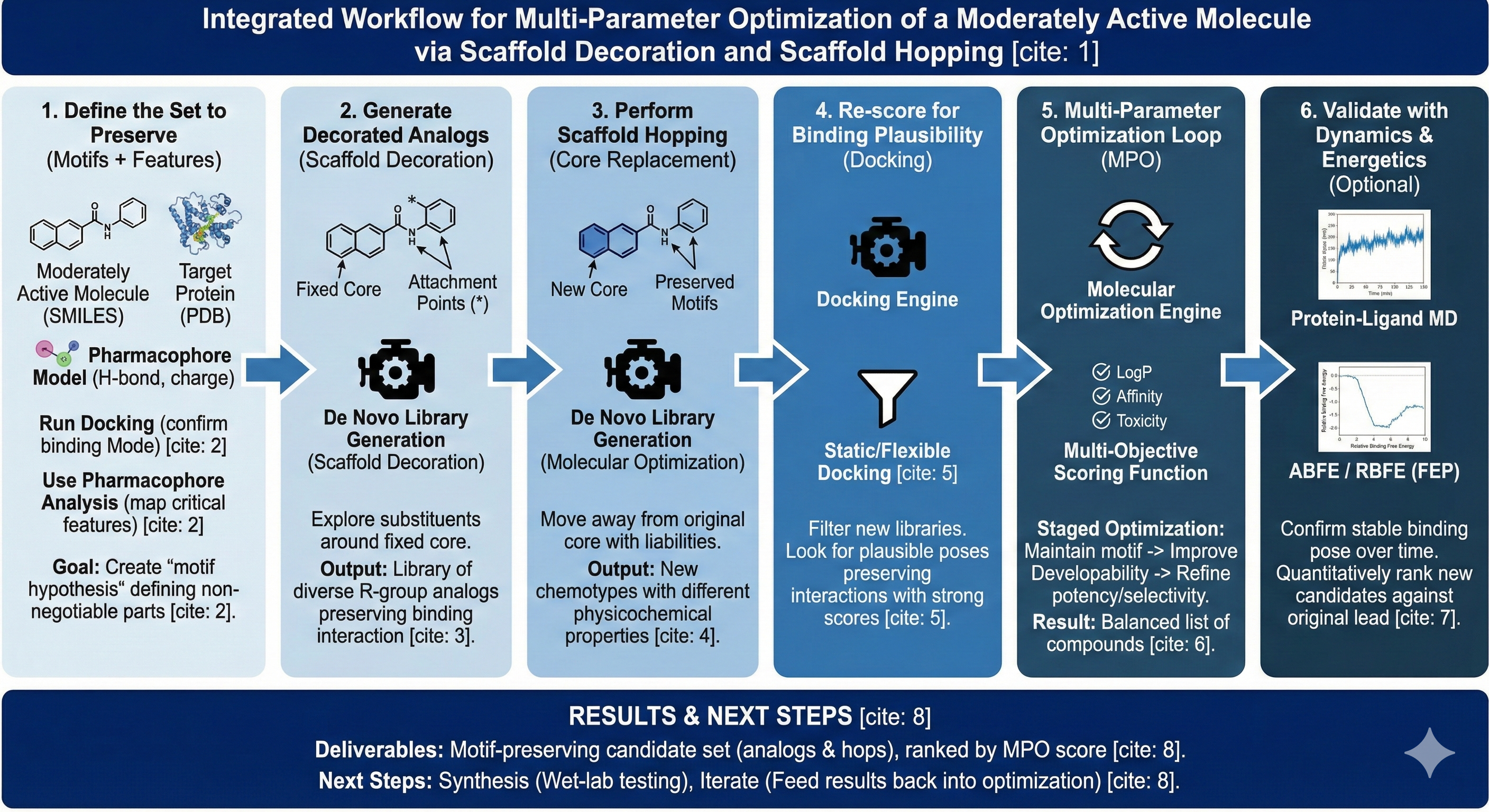
This workflow enables users to run multi-parameter optimization (MPO) using Revilico’s generative chemistry suite, specifically leveraging Scaffold Decoration and Scaffold Transformations / Diversification to propose new analogs and scaffold hops that retain key interaction motifs. The primary engine chain is: QSAR and Pharmacophore Analysis → De Novo Library Generation (Scaffold Decoration / Transformations) → Docking → Molecular Optimization (using a variety ofMPO stages). This creates a controlled loop where:
- You define what must be preserved (motifs + interaction features)
- You generate scaffold-level hypotheses that respect those constraints
- You triage structurally (docking) and iteratively improve multi-objectives (Molecular Optimization)
Required
- Starting molecule(s) as SMILES (moderately active lead(s))
- Target protein structure (experimental or predicted)
- A clear statement of what “better” means (your MPO priorities)
- You must generally know structure activity relationships of your compounds so you know what scaffolds and motifs you must preserve when running optimizations. This can be done through virtual or experimental screening and co-crystal structure determinations
- Known binding pose(s) or docking results for the starting molecule
- A description of the key motifs to preserve (functional group, ring system, H-bond pattern, charge center, etc.)
- Any known constraints or liabilities (substructures to avoid, MW limits, LogP/TPSA ranges, etc.)
- Experimental activity / selectivity data (improves scoring and QSAR usefulness)
- A set of related analogs (helps define SAR and guide similarity constraints)
- Define the Set to Preserve: Motifs + Interaction Features
- Run Docking (Static or Flexible) on the starting molecule to confirm a plausible binding mode
- Use Pharmacophore Analysis to capture the binding-critical interaction features (donor/acceptor patterns, hydrophobics, charge centers, spatial arrangement)
- Identify the motif(s) that should be preserved during decoration/hopping (e.g., hinge binder, charged anchor, aromatic stacking group)
- If experimental data is already obtained at this stage, you can re-run the compounds in docking/pharmacophore engines to gauge target engagement across certain motifs and in QSAR modeling to extract necessary substructures that drive activity.
- Generate Decorated Analogs Around the Core Scaffold
- Keep the scaffold fixed
- Specify attachment points
- Generate diverse R-group combinations that explore chemical space around your motif-preserving core
- You believe the core scaffold is correct
- You want to systematically explore substituents to improve MPO dimensions (potency + solubility + stability + etc.)
- Perform Scaffold Hopping / Core Replacement While Preserving Motifs
- Retain key interaction motifs (pharmacophore features)
- Explore alternate cores that could improve MPO properties (developability, selectivity, stability)
- You want new chemotypes (not just close analogs)
- You suspect your current core is a liability but the binding motif is correct
- Re-score for Binding Plausibility
- Start with Static Docking if the library is large
- Use Flexible Docking for a smaller set or when binding-site residue movement matters
- Plausible binding poses that preserve key interactions
- Strong scores without obvious steric clashes
- Consistency in top poses (not just one lucky pose)
- Multi-Parameter Optimization Loop
- Defining multi-objective scoring (physicochemical bounds + predicted activities + penalties)
- Using staged optimization to sequentially prioritize constraints (e.g., Stage 1: maintain motif + binding plausibility; Stage 2: improve developability; Stage 3: refine potency/selectivity)
- Balance tradeoffs (potency vs LogP vs PSA vs MW vs liabilities)
- Keep the model anchored to reasonable chemistry (avoid drifting into unrealistic structures)
- Control exploration vs conservatism using similarity settings, diversity penalties, and scoring structure
- Validate the Best Candidates with Dynamics and Energetics (Optional)
- Use Protein-Ligand MD to confirm binding stability and rule out pose artifacts
- Use ABFE/RBFE Calculation to get higher-fidelity energetic ranking within close chemical series
- A motif-preserving scaffold-decorated and scaffold-hopped candidate set
- MPO-optimized compounds ranked across potency and developability priorities
- A defensible shortlist of improved hypotheses ready for synthesis or experimental validation
- Optional physics-based validation to reduce failure risk downstream
- At this point you can continue this cycle until you have a short list of compounds that you’d like to take into synthesis. The chemical hypotheses you have should be rigorously validated computationally.
- For other parameters you’d like to optimize for, you can utilize any other Revilico Engine
Revilico makes scaffold-level MPO practical by connecting:
- motif definition (Pharmacophore Analysis)
- broad exploration (Scaffold Decoration + Scaffold Transformations)
- fast structural triage (Docking)
- deliberate multi-objective refinement (Molecular Optimization MPO stages)
- optional high-confidence validation (Protein–Ligand MD, ABFE/RBFE)