“I have a drug with demonstrated efficacy but unknown mechanism of action, and I want to identify its primary molecular target(s) to enable structure-based optimization and rational drug design”
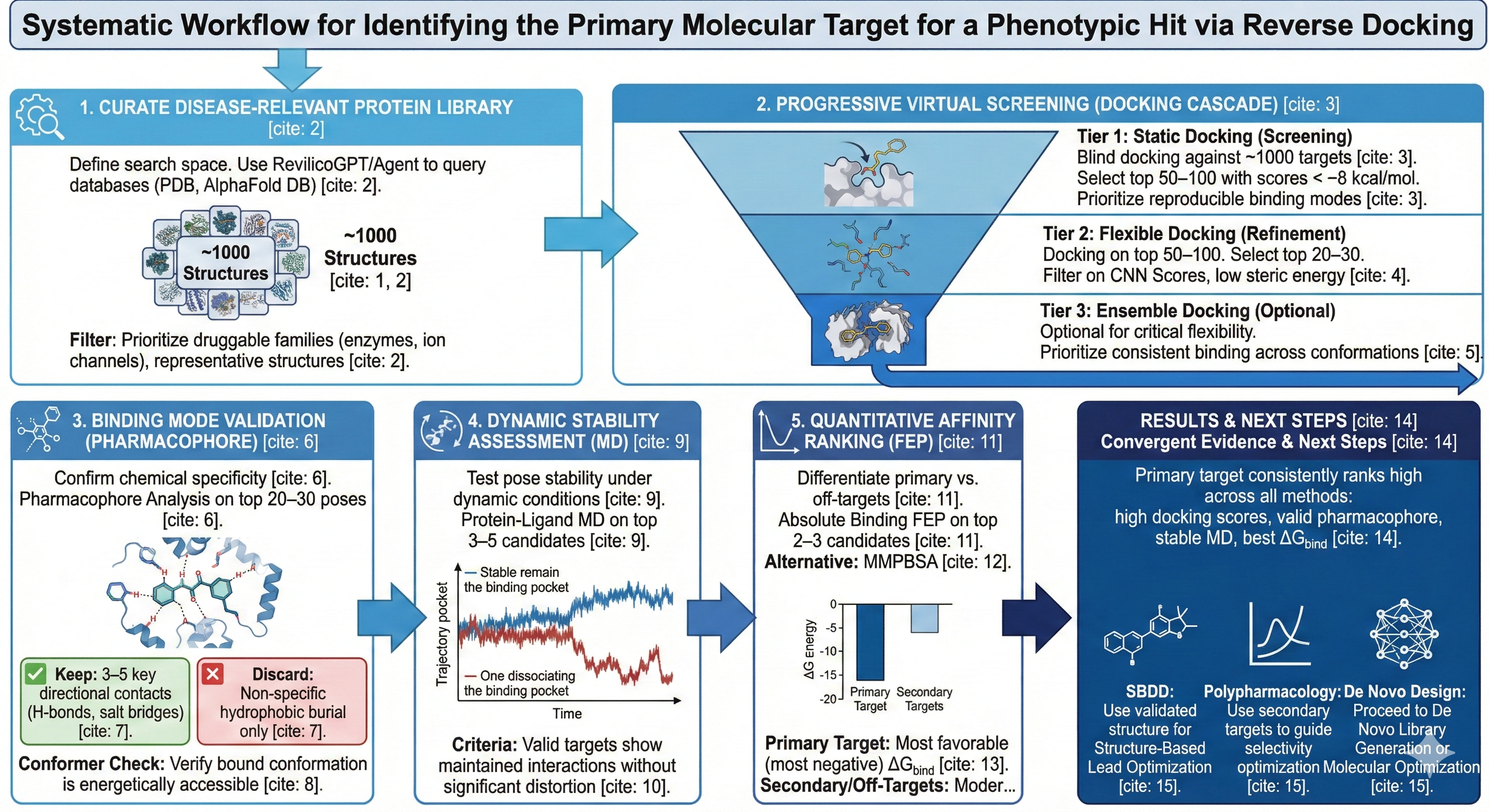
This workflow enables users to a) systematically identify primary molecular targets for phenotypic hits through reverse docking against disease-relevant protein libraries and b) validate target engagement through progressive refinement using Revico’s multi-tiered virtual screening and molecular dynamics engines. We leverage a cascade of increasingly rigorous computation methods, i.e. static docking for rapid screening, flexible docking for improved accuracy, ensemble docking for conformation sampling, protein-ligand MD for dynamic stability assessment, and free energy perturbation for quantitative affinity ranking at the highest level of accuracy. Proper target engagement validation is achieved through pharmacophore analysis that confirms reasonable binding modes and binding free energy calculations that distinguish true targets from docking artifacts. What Data Do I Need to Provide?
- Known ligand or binding partner (required for docking, MD, FEP, and Pharmacophore analysis)
- Protein Library (Optional, we have libraries on hand, or we can generate this using RevilicoGPT/Revilico Agent as well for targeted protein sets)
- Curate Disease-Relevant Protein Library
- Progressive Virtual Screening via Multi-Tiered Docking
- Validate Binding Modes with Pharmacophore Analysis
- Dynamic Stability Assessment via Molecular Dynamics
- Quantitative Affinity Ranking via Free Energy Calculations
- Ranked candidate target list with binding affinities and pose clusters
- Pharmacophore interaction maps (e.g. H-bonds, salt bridges, key contacts)
- MD stability metrics (RMSD, RMSF, interaction persistence)
- Absolute binding free energies (ΔG bind) per target
- Primary target identification with validated binding mode
- After analyzing and down-selecting your targets of interest, you can then move into generative chemistry campaigns using De Novo Library Generation, Molecular Optimization, or Custom Model training to get new libraries optimized for engagement to your target.
- After getting results from generative chemistry, you can re-score the new library using different tools to get and expand your lead series to be synthesized and tested.