“I have a moderately active molecule, and I want to optimize it for higher potency, better selectivity, and improved developability.”
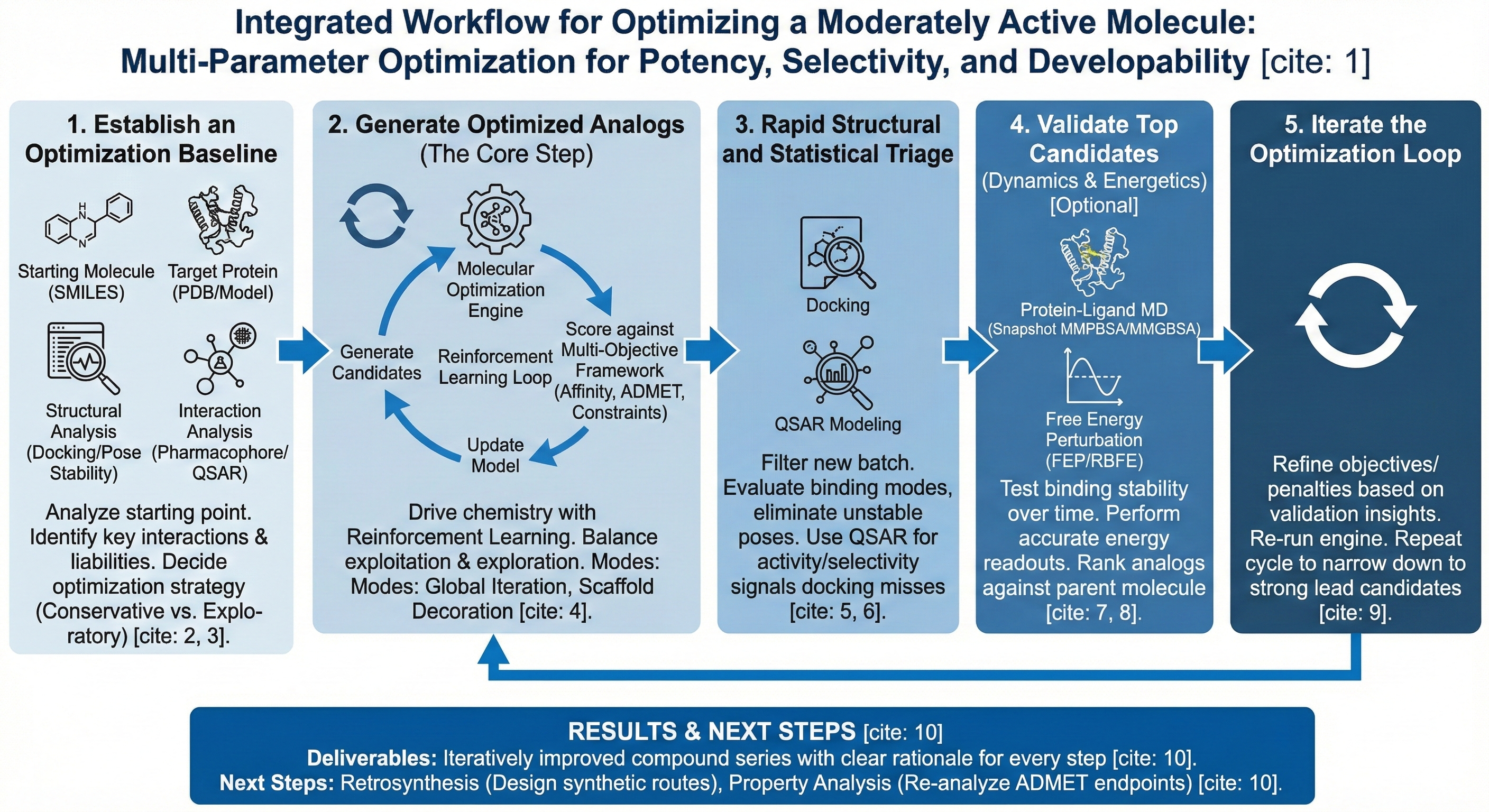
This workflow enables users to iteratively optimize a moderately active molecule using Revilico’s integrated generative, analytical, and structure-based engines, with Molecular Optimization as the core driver. At a high level, the workflow:
- Generates improved analogs of the starting molecule
- Scores and prioritizes them using fast, complementary signals
- Validates top candidates with higher-fidelity methods
- Repeats the loop as needed until clear lead candidates emerge
Required
- Starting molecule(s) as SMILES (moderately active compounds)
- Target protein structure (experimental or predicted)
- Clear optimization intent (e.g. “increase potency without increasing lipophilicity”)
- Known liabilities or constraints (avoid motifs, MW limits, polarity ranges)
- Selectivity context (off-targets or related proteins)
- Property priorities (potency vs PK vs safety tradeoffs)
- Historical activity or ADMET data (for QSAR guidance)
- Establish an Optimization Baseline
- Review Static Docking, Flexible Docking, or Ensemble Docking results (and/or experimental data) for the starting molecule to understand binding modes and pose stability
- Use Pharmacophore Analysis and QSAR Modeling to identify key interactions, liabilities, and regions of the molecule that drive activity or risk
- Decide whether optimization should be conservative (close analogs via Molecular Optimization with tight similarity constraints) or more exploratory (relaxed similarity, scaffold or substituent changes)
- Generate Optimized Analogs
- Improving predicted binding or docking performance
- Staying within desirable physicochemical ranges
- Penalizing known liabilities or unstable motifs
- Maintaining similarity to the active series (or relaxing it, if needed)
- Generating candidate molecules
- Scoring them against defined objective functions (engines that help predict parameters)
- Updating the generator model during reinforcement learning to favor better chemistry as predicted by the scoring functions.
- Rapid Structural and Statistical Triage
- Used to evaluate binding modes and relative affinity trends across static, flexible, and ensemble conditions
- Helps eliminate obvious false positives
- Provides structural intuition for SAR decisions
- Uses data-driven patterns to predict activity, selectivity, or developability signals
- Scales well across larger libraries
- Complements docking by capturing non-structural trends
- Validate Top Candidates with Dynamics and Energetics (Optional)
- Tests binding stability over biologically relevant time scales
- Reveals water effects, flexibility, and pose robustness
- Helps eliminate unstable or over-fit docking poses
- This engine is also equipped with snapshot Free Energy Perturbation (FEP) calculations using MMPSA and MMGBSA to get more accurate read outs of energies.
- Provides quantitative ranking within a focused chemical series
- Particularly useful when choosing which compounds to synthesize next
- Often used as a final filter before experimental commitment
- Is capable of highly resolving the breakdown of binding energy contributors for more resolved understanding of ligand protein engagement.
- Iterate the Optimization Loop
- Refine scoring objectives
- Adjust similarity constraints
- Introduce new penalties or priorities
- Re-run Molecular Optimization with updated guidance
- Iteratively improved compound series
- Clear rationale for why each optimization step was taken
- Reduced uncertainty before synthesis or experimental testing
- A small, prioritized set of lead-like candidates ready for the next stage
- After identifying these candidates and optimizing their activities and other properties using generative chemistry, you can move forward with Retrosynthesis to design and explore synthetic routes to utilize.
- If you’d like to re-analyze the compound set for different key properties that were also flagged for optimization, the rest of the operating system suite can be used for this as well.
Revilico enables a closed-loop optimization workflow where molecule generation, scoring, and validation are tightly integrated. Rather than relying on a single signal, users can hedge decisions across generative chemistry, structure-based modeling, and data-driven analytics, allowing optimization to move faster without sacrificing scientific control or interpretability.