“I have an identified target and a set of initial hit compounds that bind well, and I want a new library for further testing with an expanded set of chemical hypotheses.”
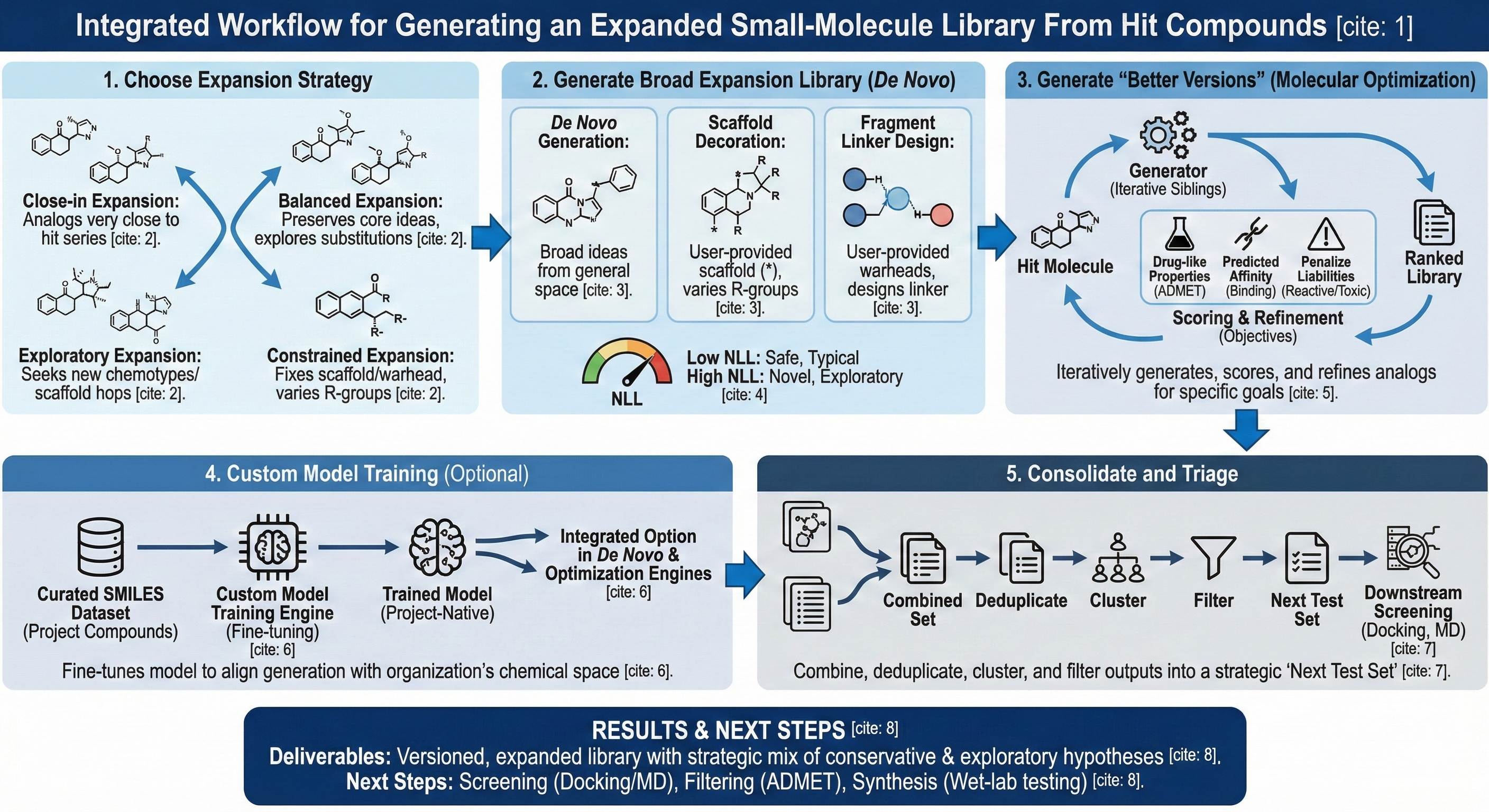
This workflow enables users to generate an expanded compound library using Revilico’s Generative Chemistry Suite, powered by reinforcement learning and a variety of scoring functions. Revilico supports three complementary ways to expand beyond your hit set:
- De Novo Library Generation, which allows you to explore new chemical space (broad expansion, scaffold hopping, fragment linking, scaffold decoration)
- Molecular Optimization generates improved “next-iteration” analogs of your hits under clear goals (property + activity objectives) for multi-parameter optimization of leads.
- Custom Model Training creates a fine-tuned generative model on your chemistry so the libraries match your project’s chemical style and constraints, allowing for optimization within pre-determined chemical spaces.
Required
- Hit compounds as SMILES (CSV or text; CSV must contain one column labeled ‘smileString’
- Your determination of your “expanded library” (close analogs vs scaffold hops vs both)
- How many new hypotheses you want (hundreds, thousands, millions)
- Known scaffolds, fragments, or warheads (if you want constrained generation)
- Preferences or constraints (size, drug-likeness, avoid substructures, etc.)
- Project-specific compound dataset (if you want custom model training to remain optimized towards your own chemical spaces)
- Choose Your Expansion Strategy
- Close-in expansion: “Give me analogs close to my hit series”
- Balanced expansion: “Keep the core ideas, but explore substitutions and related scaffolds”
- Exploratory expansion: “Find new chemotypes / scaffold hops”
- Constrained expansion: “Keep my scaffold or warheads fixed and only vary linkers / R-groups”
- Generate a Broad Expansion Library
- De Novo Generation (no starting molecules needed): broad ideas from general drug-like space biased towards certain property optimizations
- Scaffold Decoration (you provide a scaffold with * attachment points): explore R-group combinations while preserving the core
- Fragment Linker Design (you provide two warheads separated by |, with * attachment points): explore linker chemistry between fragments
Each generated molecule is accompanied by an NLL score, which helps you understand how “normal” vs “novel” the molecule is relative to the model’s learned chemistry:
- Lower NLL → safer / more typical chemistry (often good for early hit expansion)
- Higher NLL → more novel chemistry (often useful for scaffold hopping or getting unstuck from difficult performance regimes)
- Generate “Better Versions” of Your Hits
- staying within drug-like property ranges
- increasing desirability under predicted ADMET or physicochemical constraints
- encouraging novelty without breaking the series
- Optimizing for activity against specific known targets using binding affinity scoring functions
- penalizing known liabilities (reactive groups, toxic motifs)
- a ranked library of optimized hypotheses
- clear scoring summaries so you can understand why a molecule was preferred
- optional diversity controls so the library doesn’t collapse into near-duplicates
- Train a Custom Generator on Your Chemistry (Optional)
- match your chemistry patterns
- better respect your synthesis constraints
- produce compounds that “feel native” to the project based on already known structure activity relationships your chemists determined
- Consolidate Your Libraries Into a Single “Next Test Set”
- Library A (Conservative): close analogs of hit series
- Library B (Balanced): moderate exploration around scaffolds / substitutions
- Library C (Exploratory): a smaller set of scaffold hops or fragment-link ideas
- Versioned expanded compound library (new chemical hypotheses)
- A mixture of conservative and exploratory ideas (depending on your chosen strategy)
- Interpretable outputs that help you reason about novelty vs feasibility
- Libraries ready for downstream screening and prioritization
- When multiple strategies produce overlapping conclusions (e.g., similar motifs across de novo + optimization), you can move forward with greater confidence that you’re expanding in a meaningful direction.
This workflow commonly feeds into hit-to-lead prioritization steps such as:
- docking and structure-based triage
- molecular dynamics on top candidates
- ADMET filtering and multi-parameter ranking
- synthesis planning and experiment selection
- Before sending these results to the wet lab, any other Revilico engine can be used to curate compound sets with more optimal properties before spending money on synthesis or testing.
Revilico allows you to expand from hits to a next-generation test library using three complementary approaches (broad exploration, guided optimization, and project-specific model training) while keeping outputs organized, versioned, and interpretable. This enables rapid iteration without losing scientific control over what is being generated and why.