The Problem You Are Trying to Solve
“I have experimental binding or activity data, and I want to understand why these molecules behave the way they do: what interactions are driving binding, what mechanisms explain activity differences, and how this informs next design decisions.”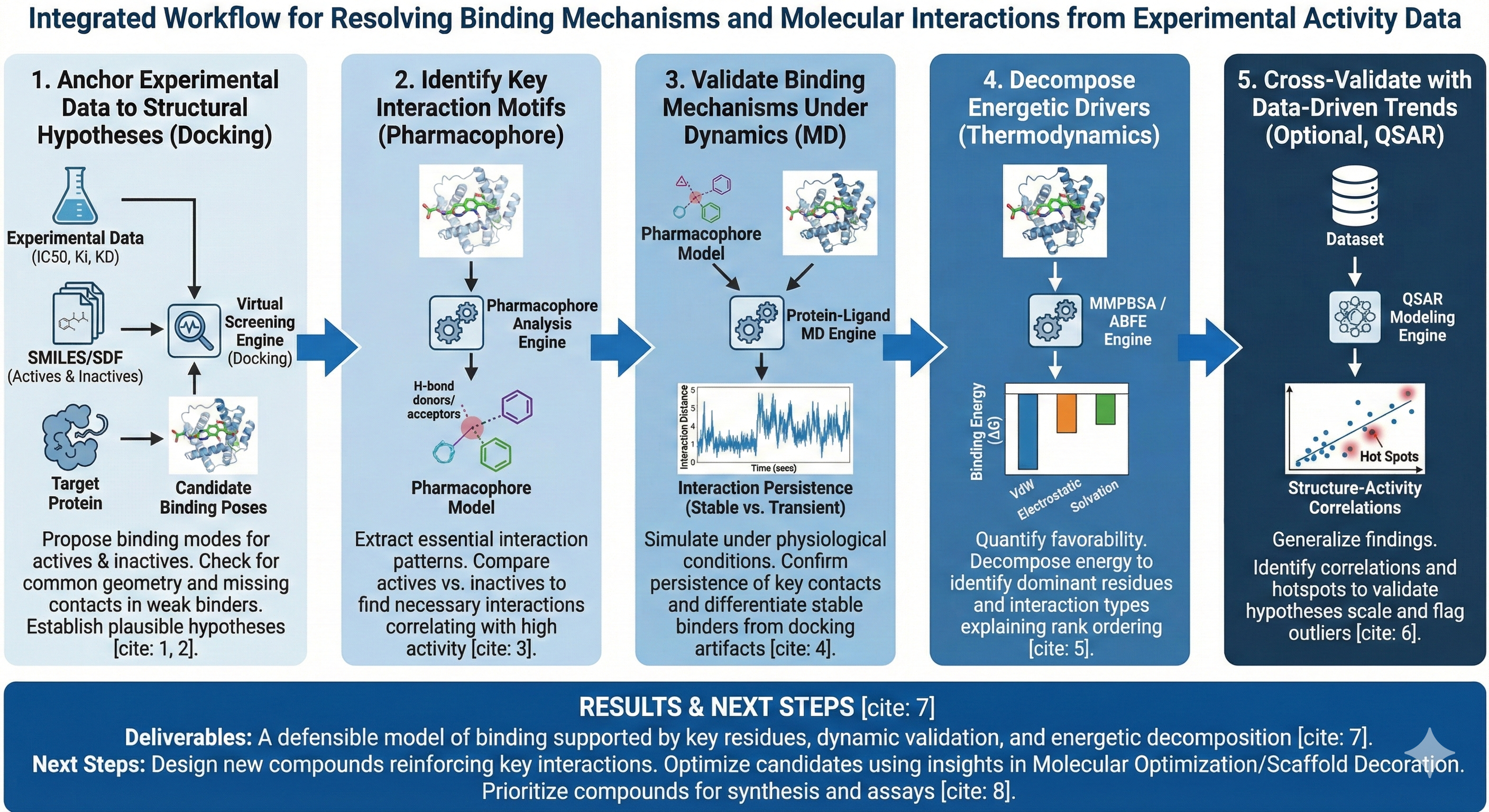
- Multiple plausible binding modes for the same ligand
- Activity trends that are difficult to rationalize from chemistry alone
- False positives or ambiguous hits without structural explanation
- Limited intuition on which interactions are essential versus incidental
Solution
This workflow uses Revilico’s binding chemistry engines to map experimental observations onto molecular interactions, combining static structure analysis, dynamic simulations, and energetic decomposition. The primary analysis chain is: Virtual Screening/Docking → Pharmacophore Analysis → Protein–Ligand MD → Energetic Decomposition (MMPBSA / ABFE where needed). This creates a layered interpretation:- Docking proposes how molecules could bind, and with what magnitude
- Pharmacophore analysis identifies what interactions matter, and provide a foundation to engineer new scaffold variants in lead optimization
- MD validates whether those interactions persist dynamically over longer time scales and in different conditions
- Energetic analysis explains why binding is favorable or unfavorable
What Data Do I Need to Provide?
Required- Experimental binding or activity data (IC₅₀, Kᵢ, Kᴅ, etc.)
- Chemical structures of tested molecules (SMILES or SDF)
- Target protein structure (experimental or predicted)
- A subset of representative compounds across activity ranges (strong, moderate, weak)
- Any known binding site information or reference ligands
- Mutagenesis data or SAR trends
- Known cofactors, ions, or binding partners
- Membrane context (if target or ligand behavior suggests it matters)
Workflow
- Anchor Experimental Data to Structural Hypotheses
Use the Virtual Screening Engine (Static,Flexible, or Ensemble) to:
- Generate candidate binding poses for active and inactive compounds
- Compare pose consistency across potency ranges
- Identify conserved versus variable interactions
- Do more active compounds share a common binding geometry?
- Do weak binders fail to make key contacts or show unstable poses?
- Identify Key Interaction Motifs
- Extract hydrogen bond donors/acceptors, hydrophobic features, aromatic interactions, and charge centers
- Compare pharmacophores across active vs inactive compounds
- Identify interaction features that correlate with experimental activity
- Which interactions appear necessary for activity?
- Which regions tolerate variation?
- Are there missing interactions explaining weak activity?
- What potential modifications can be made for later stage lead series expansion?
- Validate Binding Mechanisms Under Dynamics
Use Protein-Ligand MD to:
- Test whether docked poses remain stable over time
- Observe interaction persistence (H-bonds, salt bridges, hydrophobic contacts)
- Identify conformational rearrangements or ligand drift
- Compare dynamics between high- and low-activity compounds
- Decompose Energetic Drivers of Binding
Use:
- MMPBSA / MMGBSA for fast energetic breakdown across MD trajectories
- ABFE selectively when absolute binding favorability must be quantified
- Decompose van der Waals, electrostatic, and solvation contributions, among other energetic components.
- Identify residues or interactions dominating binding energetics, and how these change over time scales
- Explain experimental rank ordering in energetic terms
- Cross-Validate with Data-Driven Trends (Optional)
- Identify structure–activity correlations and hot spots of activity within chemical space
- Validate whether interaction hypotheses scale across chemical space
- Flag outliers or inconsistent data points
- Experimental datasets are large
- Multiple binding modes may exist
Results
- A clear, mechanistic explanation of experimental binding/activity data
- Identified key residues and interaction motifs driving activity
- Dynamic validation of binding hypotheses
- Energetic decomposition supporting observed trends
- A defensible model of how and why molecules bind
Now What? I understand the binding mechanism, but what’s next?
Typical next steps include:- Designing new compounds that reinforce key interactions
- Eliminating false positives before hit expansion
- Feeding insights into generative chemistry on the platform using Molecular Optimization or Scaffold Decoration engines to create new molecular hypotheses
- Prioritizing compounds for synthesis or further biophysical assays
Why Revilico?
Revilico enables experimental data interpretation by connecting:- Structural hypotheses (Docking)
- Interaction abstraction (Pharmacophore Analysis)
- Physical realism (Protein–Ligand MD)
- Quantitative energetics (MMPBSA / ABFE)