“I have a molecular target without obvious or stable binding sites, and I want to identify druggable pockets suitable for structure based drug design”
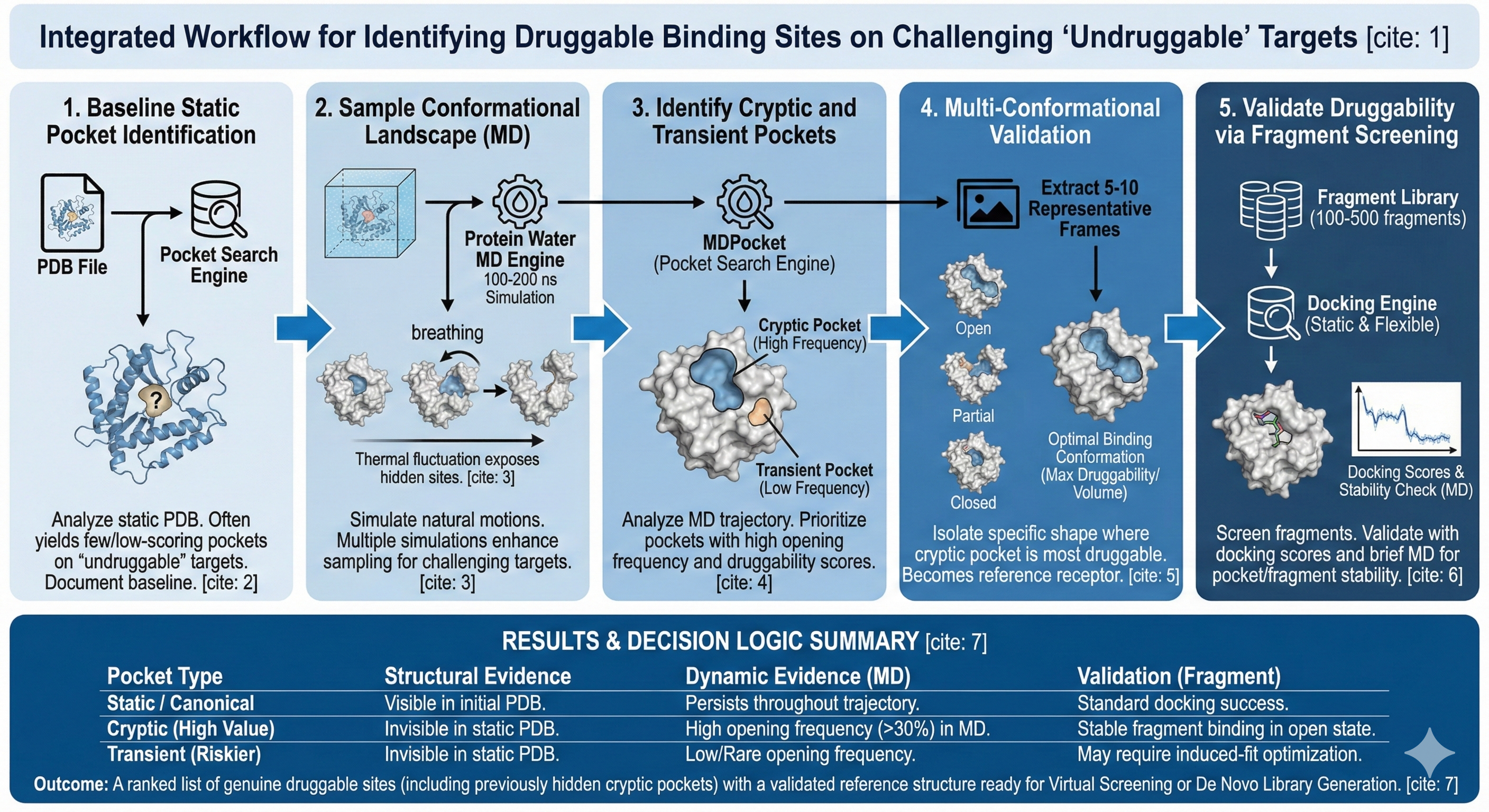
This workflow enables users to a) systematically identify all potential binding sites on challenging targets through static pocket analysis and b) discover cryptic, transient, and allosteric pockets that only appear during protein motion using molecular dynamics-based pocket detection across Revilio’s structural analysis engines. Proper druggability assessment is achieved by analyzing pocket properties across multiple protein states and ranking sites by persistence, accessibility, and druggability scores, helping eliminate status analysis blind spots and increasing confidence that identified pockets represent genuine druggable sites even on targets traditionally considered undruggable. What Data Do I Need to Provide?
- PDB File of your protein structure (will be used for Pocket Search, Docking, and MD simulations)
- Baseline Static Pocket Identification
- Sample Conformational Landscape via Molecular Dynamics
- Identify Cryptic and Transient Pockets
- Multi Conformational Validation
- Validate Druggability with Fragment Screening
- Comprehensive pocket inventory with druggability scores and volumes
- Temporal pocket profiles showing opening frequency and persistence across MD simulations
- Representative protein conformations with pocket open/closed states
- Fragment screening validation results
- Ranked druggability site recommendations with strategic classifications
- Utilize the Virtual Screening engine or De Novo Library Generation to begin your campaign and identify initial hits. Utilizing the knowledge gained from pocket identification, you can target your docking calculations on the box of choice and across conformational states to ensure you’re representing the biological system properly.
This workflow addresses the challenge of identifying druggable sites on targets without obvious or stable binding pockets by combining static analysis with dynamic molecular dynamics (MD) simulations. It systematically discovers cryptic and transient binding sites through MD and MDPocket, then validates the most persistent and druggable pockets using fragment screening to establish genuine targets for downstream structure-based drug design.