The Problem You Are Trying to Solve
“I want to run large-scale separation-of-function mutants on my target to validate ligand–protein interactions, but experimentally generating and testing all variants is too intensive.”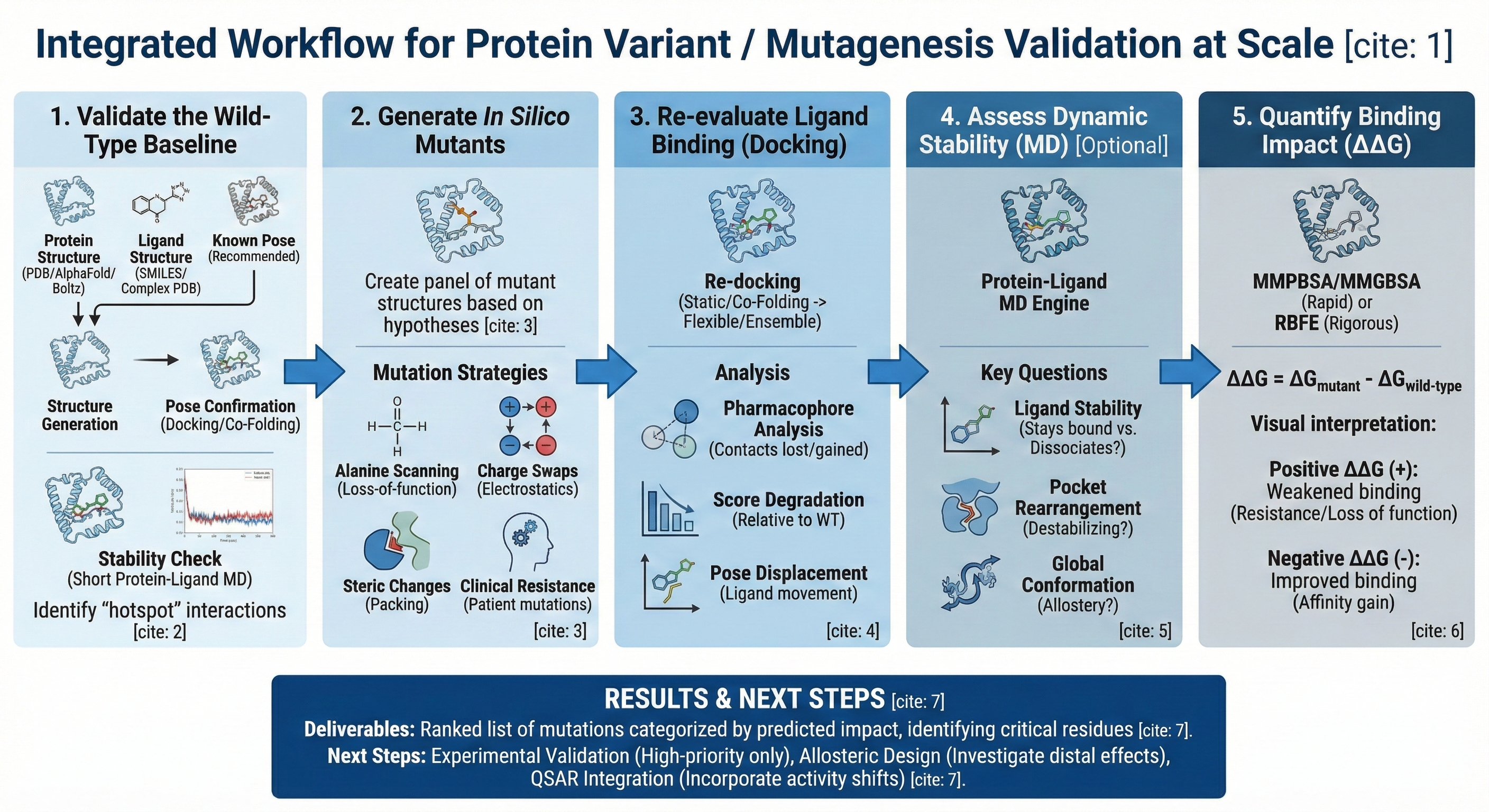
- Validating binding site hypotheses
- Confirming key interaction residues
- Distinguishing orthosteric vs allosteric mechanisms
- Understanding resistance mechanisms
This workflow simulates mutational effects on binding affinity, pose stability, and protein conformational behavior before any wet-lab investment. The primary workflow chain is as follows: Baseline Complex Validation → In Silico Mutant Generation → Docking Re-evaluation → MD Stability Assessment → ΔΔG / Free Energy Comparison → Ranked Variant Prioritization. This enables users to computationally predict the effects of amino acid changes on small-molecule binding, allowing you to prioritize which variants to generate experimentally.
What Data Do I Need to Provide?
Required- Protein structure (PDB or predicted structure)
- Ligand structure (SMILES or complex PDB)
- Known binding pose (co-crystal or validated docked model)
- List of residues of interest that contribute to biological activities or ligand engagement (e.g., predicted binding site residues)
- Known resistance mutations which can be based on pure biophysical analysis of the protein or based on genomic patient data to test for certain amino acid substitutions that exist in the general patient populations.
- Functional assay results for calibration
Workflow
- Validate the Wild-Type Baseline
- Generate the baseline structure using the amino acid only across AlphaFold, OpenFold, and Boltz structure generation engines
- Confirm ligand binding pose with Docking or Co-Folding models
- Validate stability with short Protein–Ligand MD
- Identify key hydrogen bonds, electrostatic contacts, hydrophobic interactions and how they change as a function of time.
- Generate In Silico Mutants
- Select residues for mutagenesis (e.g., binding site, distal regulatory regions)
- Define mutation types (alanine scan, conservative substitutions, resistance-inspired variants)
- Generate mutant protein structures computationally using our variety of protein structure generation tools
- Amino acids that are known drivers of binding and activity. Mutations can allow confirmation of key binding modes and energies.
- Alanine scanning (loss-of-function mapping)
- Charge swaps (e.g., Asp → Lys)
- Size/steric changes (e.g., Phe → Ala)
- Known clinical resistance mutations
- Re-evaluate Ligand Binding via Docking
- Re-dock the ligand into the altered structure with Static Docking or Co-Folding as a first-pass
- Compare predicted binding affinity and pose shifts, and you can confirm certain binding poses and amino acid engagements using the Pharmacophore Analysis Engine.
- Repeat with Flexible Docking if local rearrangement is expected, or with Ensemble docking for more sophisticated and dynamic protein systems.
- Loss or gain of critical contacts
- Pose displacement
- Score degradation relative to wild-type protein mutants
- Assess Dynamic Stability (Optional)
- Protein–Ligand MD for high-impact mutants
- Compare RMSD, RMSF, SASA, and interaction persistence against wild-type
- Does the ligand remain stably bound?
- Does the mutation induce destabilizing pocket rearrangement?
- Does distal mutation alter global conformational behavior?
- Quantify Binding Impact
- Use MMPBSA or MMGBSA on Protein Ligand MD for rapid ΔG comparison
- Use RBFE to compute ΔΔG between wild-type and mutant complexes
- Use ABFE for absolute binding comparison (if needed)
- ΔΔG = ΔG_mutant − ΔG_wild-type
- Negative ΔΔG → improved binding
- Positive ΔΔG → weakened binding
Results
- Ranked mutation list by predicted impact on binding
- Identified critical binding residues
- Separation-of-function candidate mutants
- Energetic and structural rationale for each mutation
- Focused experimental validation
- Reduced mutagenesis burden
- Mechanistic clarity for ligand engagement
Integration with Other Engines (Optional)
Depending on study goals, this workflow can integrate with:- Allosteric Design workflows (if distal mutations alter regulatory pockets)
- QSAR Modeling (incorporate mutation-dependent activity shifts)
- ADMET-AI (if mutations influence ligand orientation and downstream design)
- scRNA-Seq Analysis (if variant alters pathway activation)
- Quantum Chemistry engines (if mutation changes electrostatic environment significantly)
- You can utilize this information to create more informed protein variants of your validated complexes that you’d like to evaluate for separation of function mutagenesis.
- Creating the right complexes can allow you to confirm structural hypotheses of binding, and to ensure that your hypotheses of what amino acids drive binding are correct
- You can utilize a variety of different engines on the dashboard to test your new protein variants for structural similarity, or your new complexes in more advanced scoring engines.
Why Revilico?
Revilico enables scalable mutational validation by combining:- Structural modeling of variants
- Docking-based interaction reassessment
- Dynamic stability simulation
- Thermodynamic free energy comparison
- Unified visualization and interpretation